Analysis of Cancer Stroma Interface in Xenium#
%load_ext autoreload
import os
import sys
import time
import logging
import re
from tqdm import tqdm
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
from PIL import Image
import json
import anndata as ad
import scanpy as sc
from natsort import natsorted
import numpy as np
np.random.seed(42)
# import itables
# from itables import show
# itables.init_notebook_mode(all_interactive=True)
sys.path.append(os.path.dirname(os.getcwd()))
import seaborn as sns
from gridgene import get_arrays as ga
from gridgene import contours, get_masks
# from GRIDGEN.src.mask_properties2 import GetMasksProperties
from gridgene.mask_properties2 import MaskAnalysisPipeline, MaskDefinition
# define the logger : can be None, and is set to INFO
# Custom logger setup
logger = logging.getLogger('contour_logger')
handler = logging.StreamHandler()
formatter = logging.Formatter('%(asctime)s - %(name)s - %(levelname)s - %(message)s')
handler.setFormatter(formatter)
logger.addHandler(handler)
logger.setLevel(logging.INFO)
Get the files for Xenium
tma1_path = '../../xenium_data/HLA/GD_TMA1_S3/fov_filtered'
files_tma1 = os.listdir(tma1_path)
to_exclude = [
'TMA1_Selection14_filtered.csv' , # little tumour
'TMA1_Selection15_filtered.csv', # tonsil
'TMA1_Selection18_filtered.csv' , # normal
'TMA1_Selection24_filtered.csv', # tonsl
'TMA1_Selection27_filtered.csv', # low quality
'TMA1_Selection32_filtered.csv', # low quality
'TMA1_Selection33_filtered.csv', # low quality
]
files = [os.path.join(tma1_path, file) for file in files_tma1 if file not in to_exclude]
print(len(files))
metadata_file = '../../xenium_data/HLA/map_HLA_19_09_2024.xlsx'
metadata = pd.read_excel(metadata_file)
metadata['name'] = metadata['XENIUM_TMA'] + '_' + metadata['Selection'].str.replace(' ', '')
metadata = metadata.loc[metadata["XENIUM_TMA"] == "TMA1"]
metadata.head()
20
| XENIUM_TMA | Selection | annotation | Hes | TMA | COSMX selection | Case | Block | Unnamed: 8 | MMR | HLA | B2M abcam | IHC group | B2M gene defect | Final group | name | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | TMA1 | Selection 7 | NaN | yes | GD_TMA1 | COSMX | 17/H15722 | 1I | S00159 | MMRd | negative | mut pattern | HLA defect | no mutation | HLA defect | TMA1_Selection7 |
| 1 | TMA1 | Selection 8 | NaN | yes | GD_TMA1 | COSMX | 17/H15137 | 1G | S00154 | MMRd | positive | positive | HLA+ | no mutation | HLA+ | TMA1_Selection8 |
| 2 | TMA1 | Selection 9 | NaN | yes | GD_TMA1 | COSMX | 17/H09934 | 1M | S00120 | MMRd | positive | positive | HLA+ | NaN | HLA+ | TMA1_Selection9 |
| 3 | TMA1 | Selection 11 | NaN | yes | GD_TMA1 | COSMX | 17/H08334 | 1E | S00102 | MMRd | negative | positive | HLA defect | mutation (exon 2) | B2M defect | TMA1_Selection11 |
| 4 | TMA1 | Selection 12 | NaN | yes | GD_TMA1 | COSMX | 17/H07092 | 1AD | S00096 | MMRd | cytoplasmic | negative | B2M defect | mutation (exon 1) | B2M defect | TMA1_Selection12 |
Derive cancer and stroma masks
Apply GRIDGEN to Xenium to derive cancer and stroma masks (similar to the previous notebook: “Cancer Stroma in Xenium”).
After this, expansions from the cancer mask inside the stroma mask are derived, and mask information is retrieved.
We will show this in one example first and then apply to the cohort.
# param tum
target_tum = ['EPCAM', 'SMIM22','CLDN3', 'KRT18','LGALS4', 'KRT8', 'ELF3','TSPAN8', 'STMN1', 'CD47', 'MYC', 'LGALS3']
density_th_tum = 10 # 50 30
min_area_th_tum = 700 #1000 check how much is a cell
kernel_size_tum = 10 # 20
# param empty
density_th_empty = 40
min_area_th_empty = 400 #400
kernel_size_empty = 10
file_csv = files[9]
df_total = pd.read_csv(file_csv)
df_total = df_total[['x_location', 'y_location', 'feature_name']]
df_total = df_total.rename(columns={'feature_name': 'target'})
df_total = df_total[~df_total['target'].str.contains('System|egative')]
df_total['X'] = df_total['x_location'] - min(df_total['x_location'])
df_total['Y'] = df_total['y_location'] - min(df_total['y_location'])
n_genes = len(df_total['target'].unique())
height = int(max(df_total['X'])) + 1
width = int(max(df_total['Y'])) + 1
print('df shape', df_total.shape)
print('shape',height,width,n_genes)
# this makes the sparse df to an array with the spatial information
target_dict_total = {target: index for index, target in enumerate(df_total['target'].unique())}
array_total = ga.transform_df_to_array(df = df_total, target_dict=target_dict_total, array_shape = (height, width,len(target_dict_total))).astype(np.int8)
# creating subsets
df_subset_tum, array_subset_tum, target_indices_subset_tum = ga.get_subset_arrays(df_total, array_total,target_dict_total,
target_list=target_tum, target_col = 'target')
# obtain contours
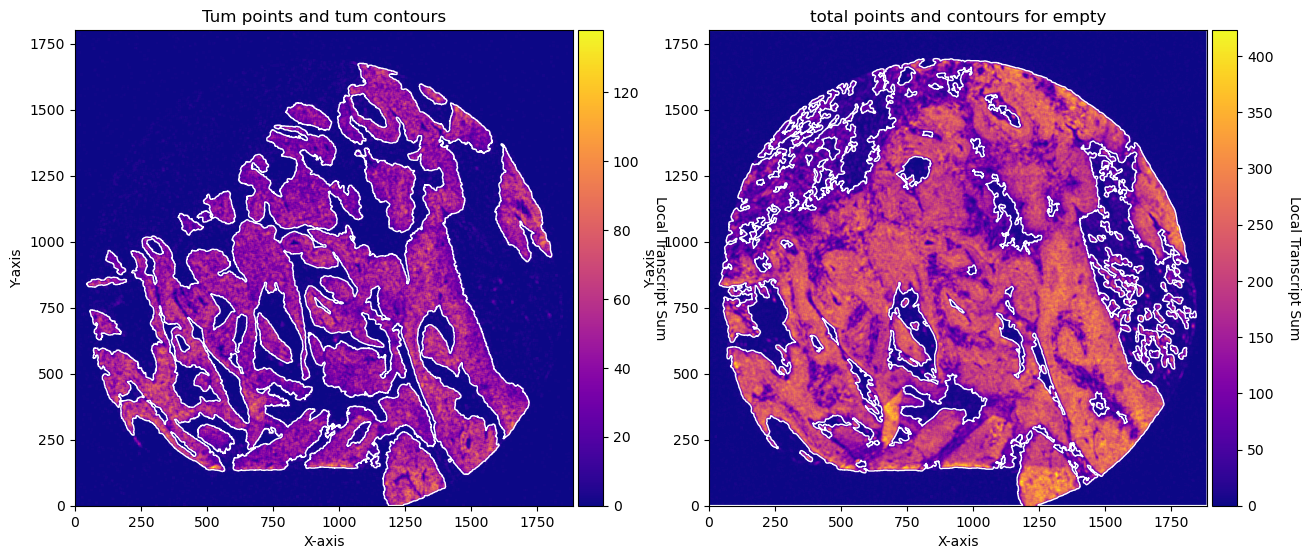
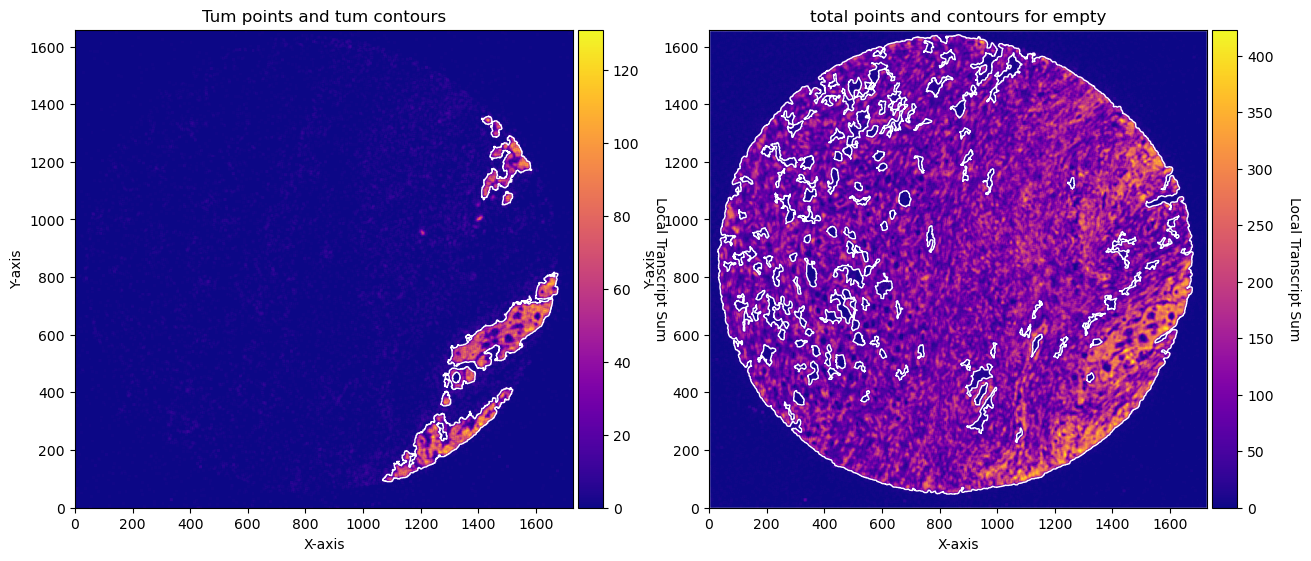
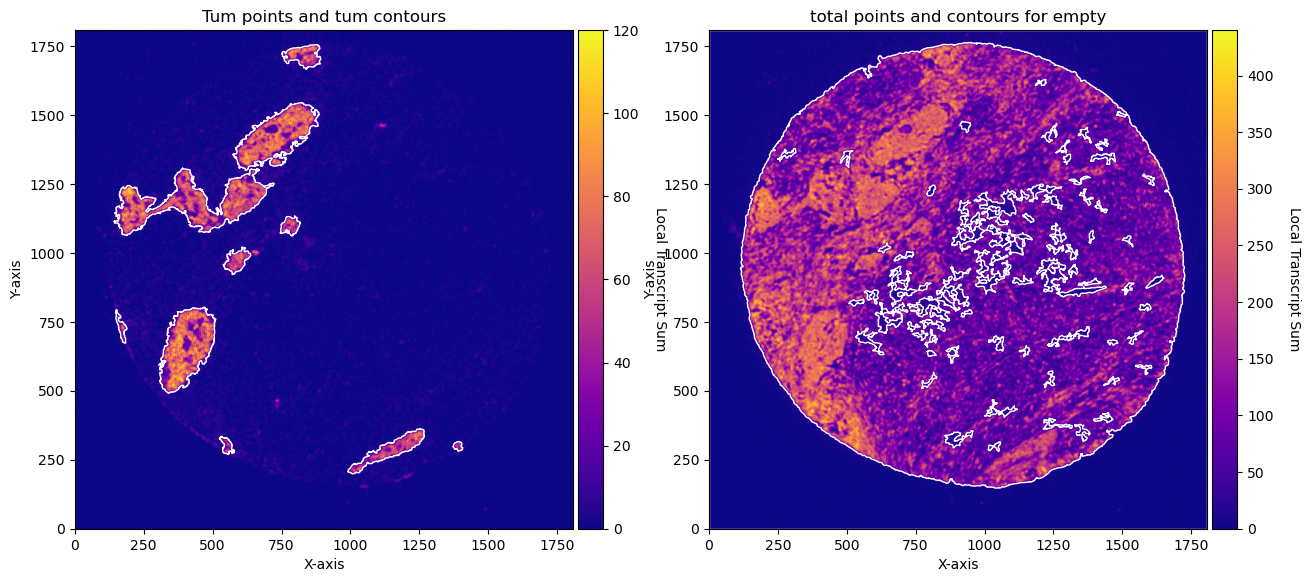
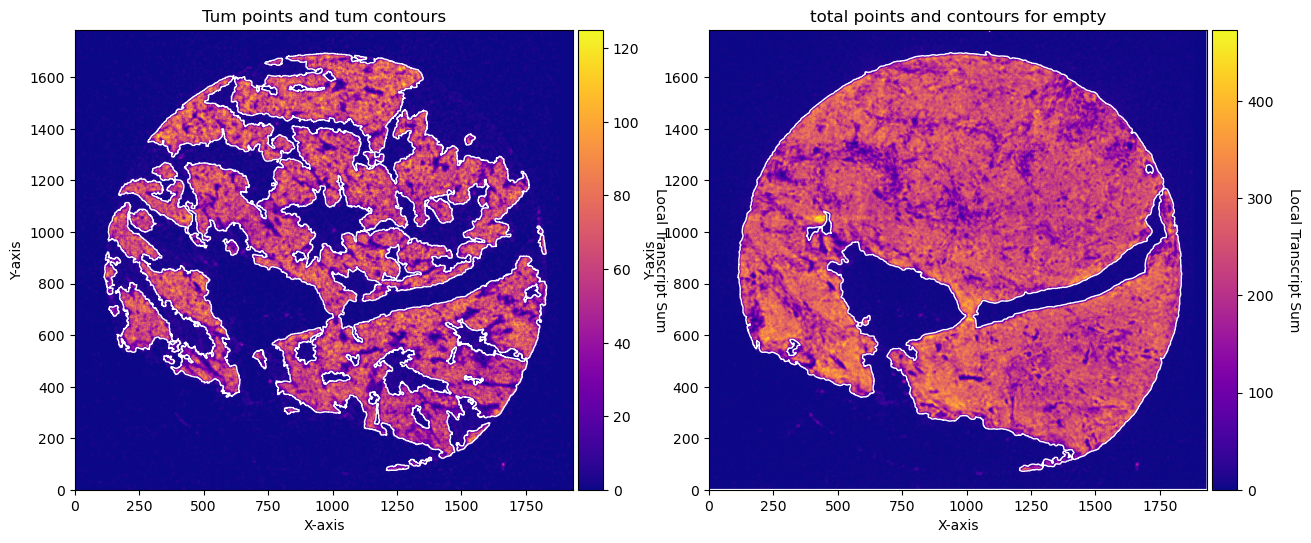
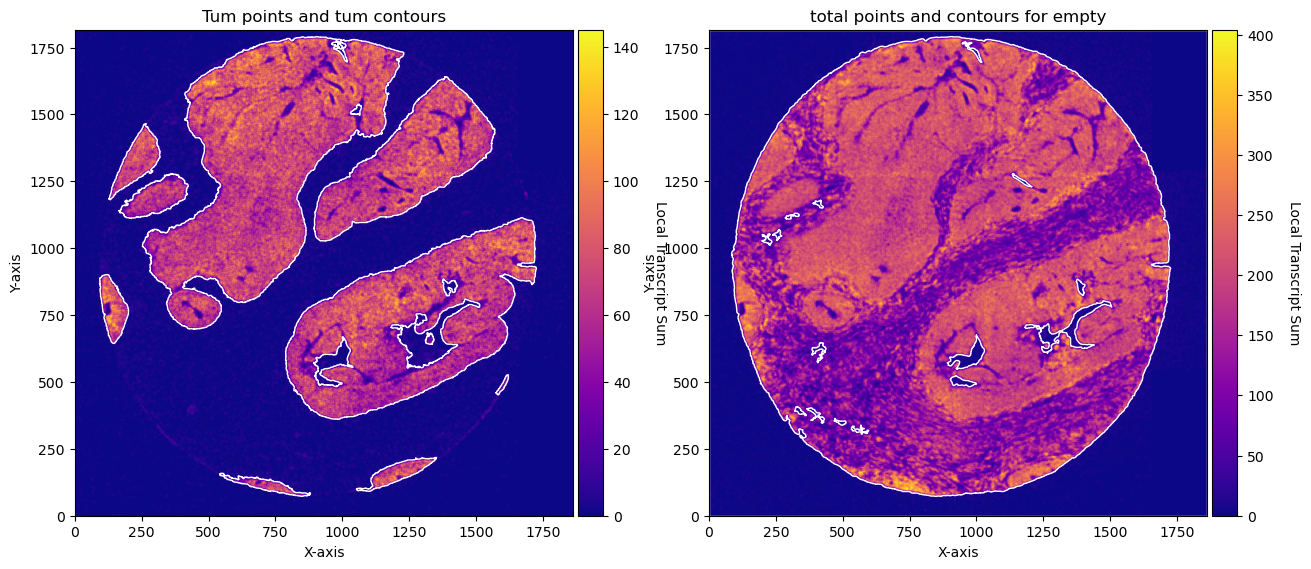
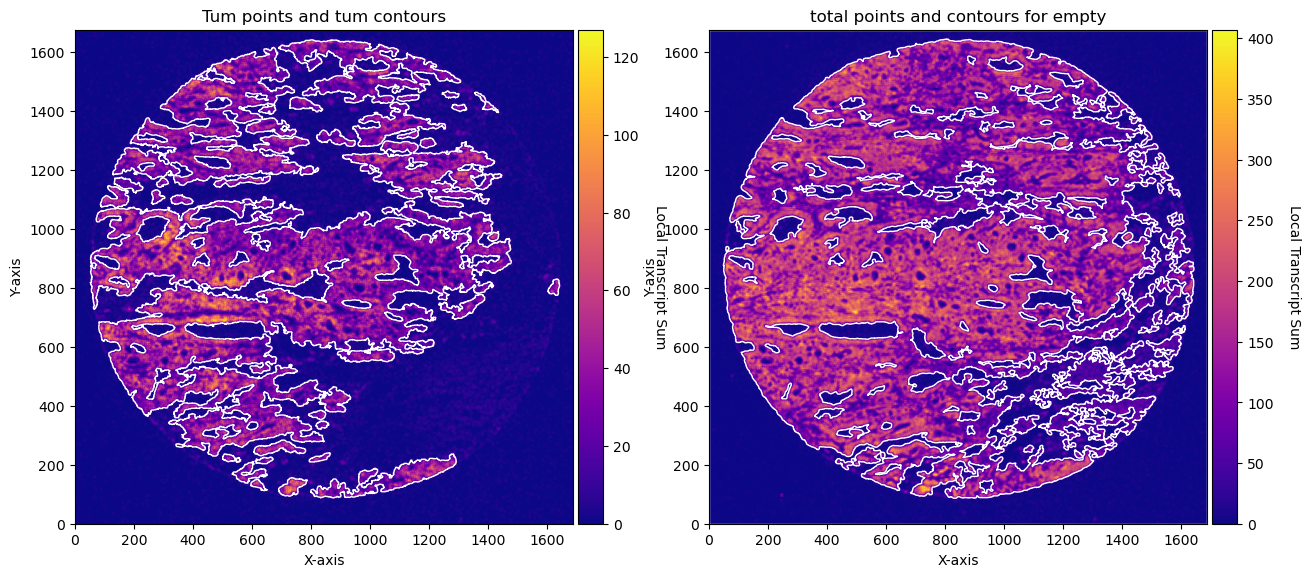
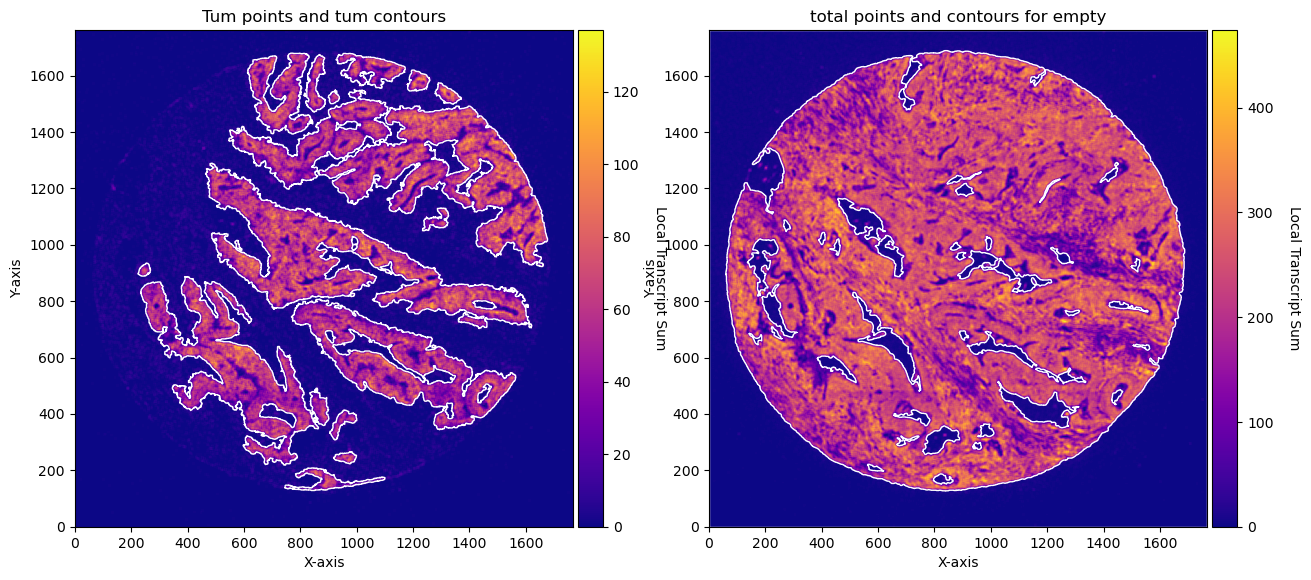
CTum = contours.ConvolutionContours(array_subset_tum, contour_name='tum')
CTum.get_conv_sum(kernel_size=kernel_size_tum, kernel_shape='square')
CTum.contours_from_sum(density_threshold = density_th_tum,
min_area_threshold = min_area_th_tum , directionality = 'higher')
CEmpty = contours.ConvolutionContours(array_total, contour_name='empty')
CEmpty.get_conv_sum(kernel_size=kernel_size_empty, kernel_shape='square')
CEmpty.contours_from_sum(density_threshold = density_th_empty,
min_area_threshold = min_area_th_empty, directionality = 'lower') # attention that directionality is lower here
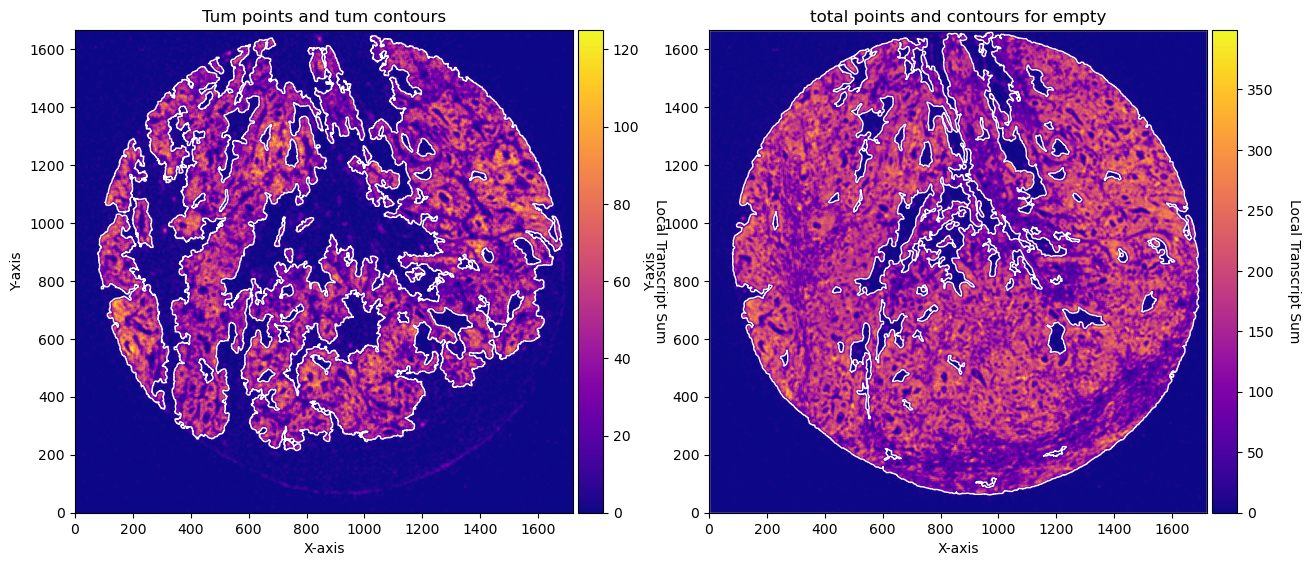
# PLOTs -- instead of plotting just with the function for example: CTum.plot_contours_scatter, we will make a composite image
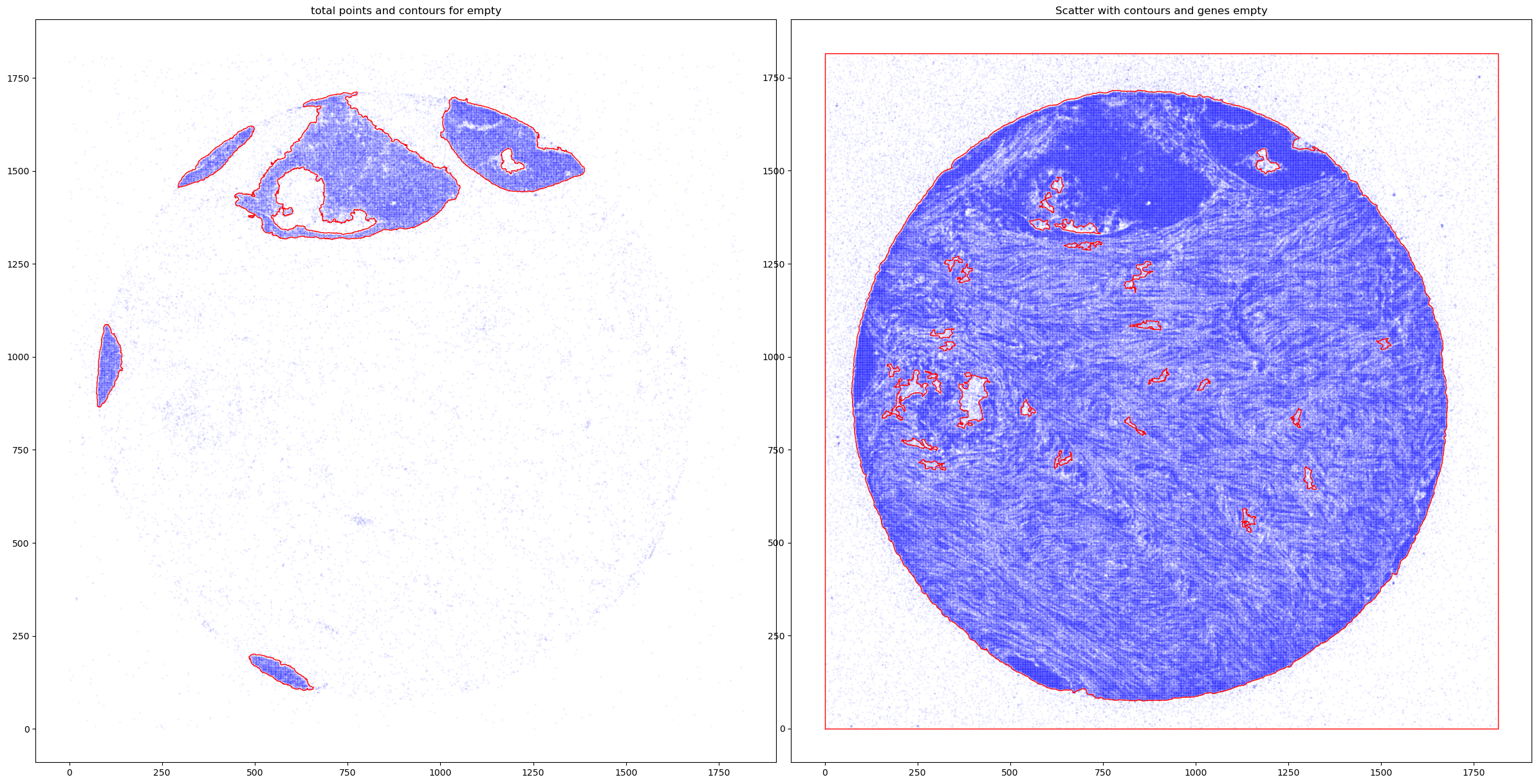
fig, axs = plt.subplots(1, 2, figsize=(30, 15))
CTum.plot_contours_scatter(path=None, show=False, s=0.05, alpha=0.5, linewidth=1,
c_points= 'blue',c_contours= 'red', ax=axs[0])
axs[0].set_title('Tum points and tum contours')
CEmpty.plot_contours_scatter(path=None, show=False, s=0.05, alpha=0.5, linewidth=1,
c_points= 'blue',c_contours= 'red', ax=axs[1])
axs[0].set_title('total points and contours for empty')
plt.subplots_adjust(wspace=0.02) # Adjust to your preference
plt.show()
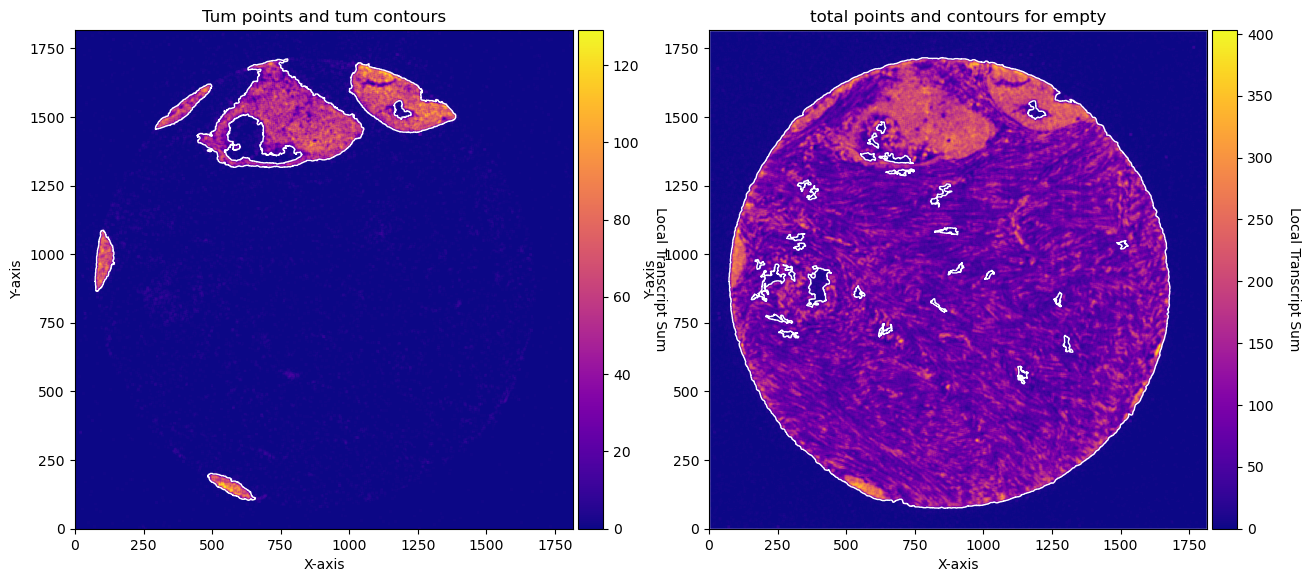
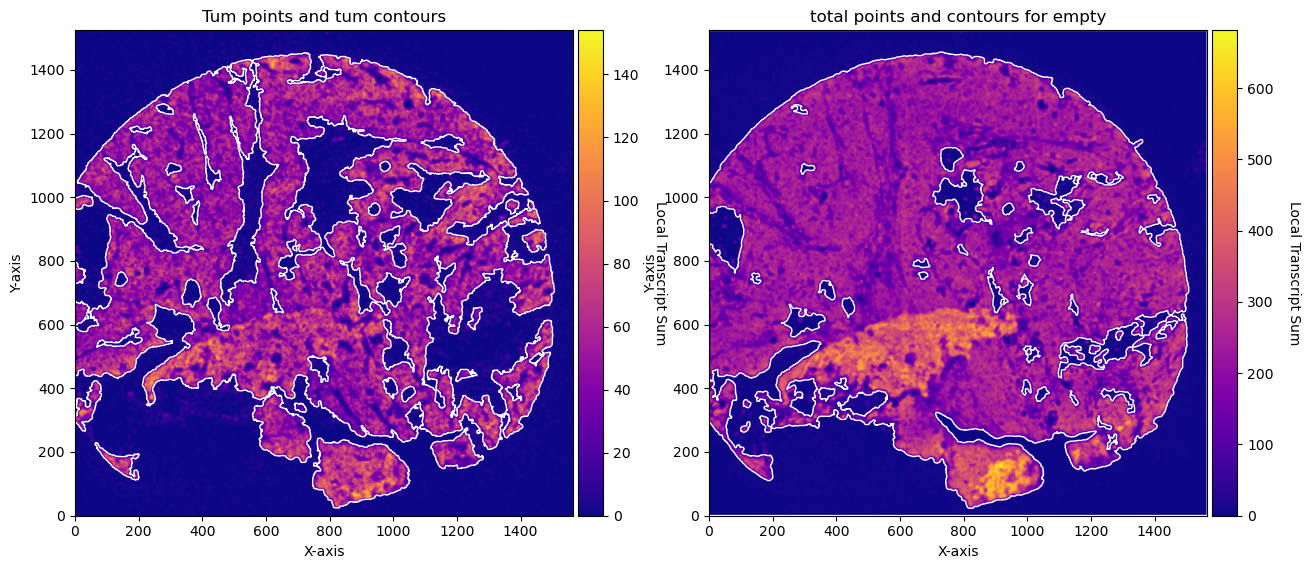
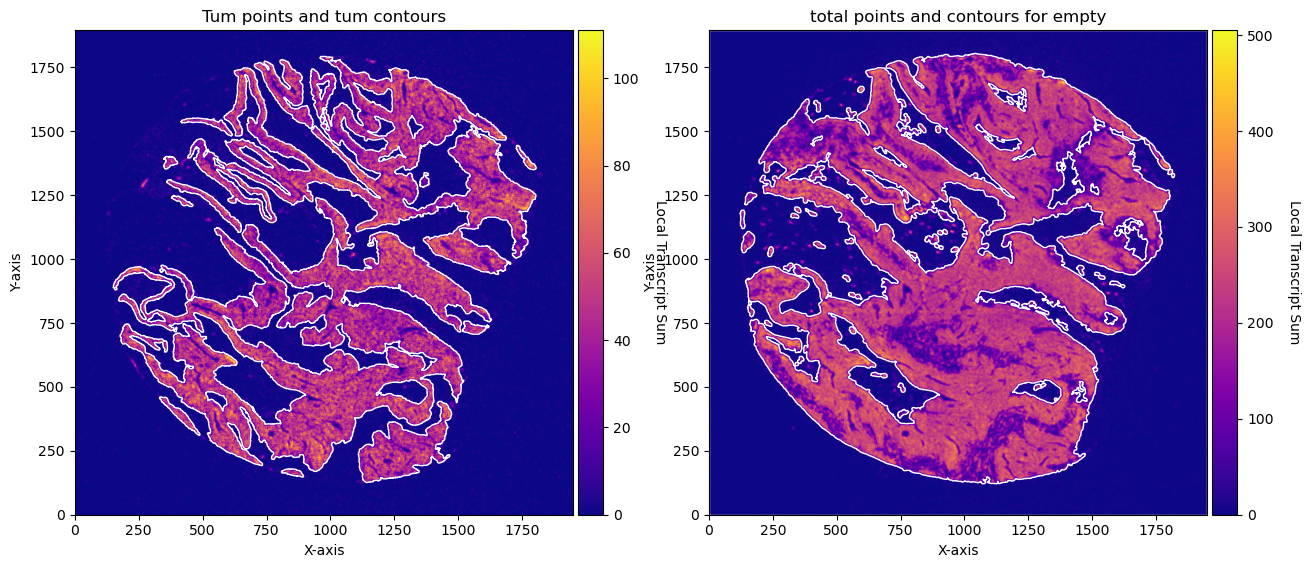
fig, axs = plt.subplots(1, 2, figsize=(15, 10))
CTum.plot_conv_sum(cmap='plasma', c_countour='white', ax=axs[0])
axs[0].set_title('Tum points and tum contours')
CEmpty.plot_conv_sum(cmap='plasma', c_countour='white', ax=axs[1])
axs[1].set_title('total points and contours for empty')
plt.show()
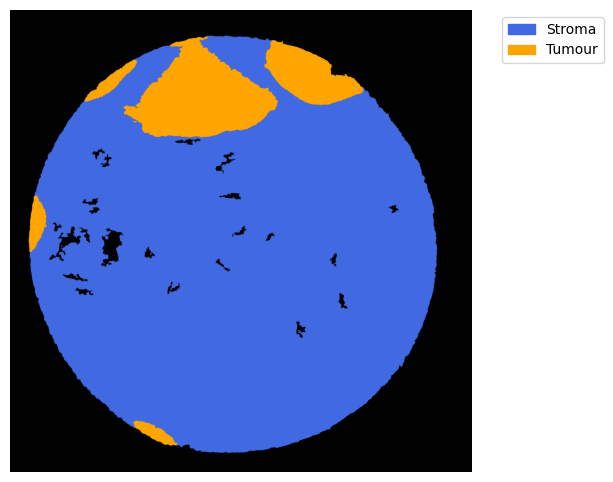
#### obtain masks
GM = get_masks.GetMasks(image_shape = (height, width))
mask_empty = GM.create_mask(CEmpty.contours)
mask_tum = GM.create_mask(CTum.contours)
mask_tum = GM.fill_holes(mask_tum)
mask_stroma = GM.subtract_masks(np.ones((height, width), dtype=np.uint8), mask_tum, mask_empty)
mask_stroma = GM.filter_binary_mask_by_area(mask_stroma, min_area=700)
GM.plot_masks(masks=[mask_stroma, mask_tum], mask_names=['Stroma', 'Tumour'],
background_color=(1, 1, 1), mask_colors={'Stroma': (65, 105, 225), 'Tumour': (255, 165, 0)},
path=None, show=True, ax=None, figsize=(8, 6))
df shape (2084362, 5)
shape 1817 1817 480
2025-06-13 15:12:09,584 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:12:09,723 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 7
2025-06-13 15:12:09,723 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0801 seconds
contours_from_sum took 0.0590 seconds
get_conv_sum took 0.5682 seconds
2025-06-13 15:12:10,869 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 29
contours_from_sum took 0.5778 seconds
2025-06-13 15:12:12,882 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:12:12,901 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
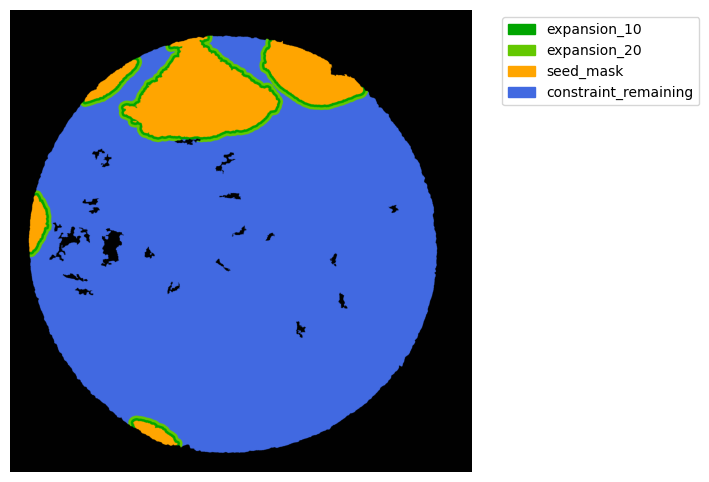
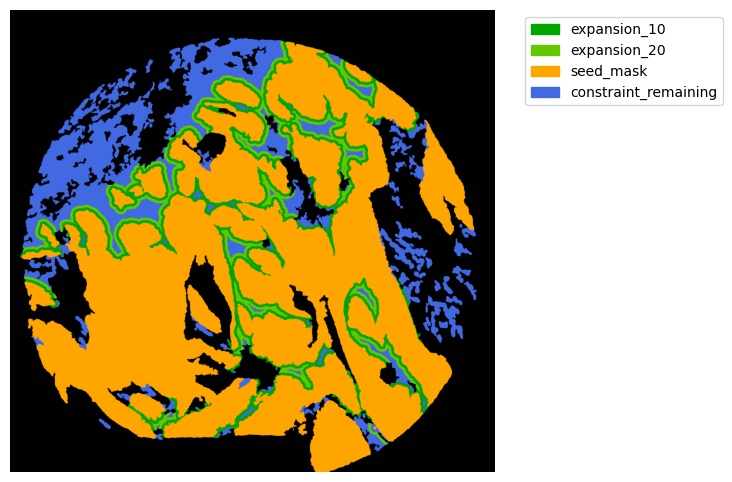
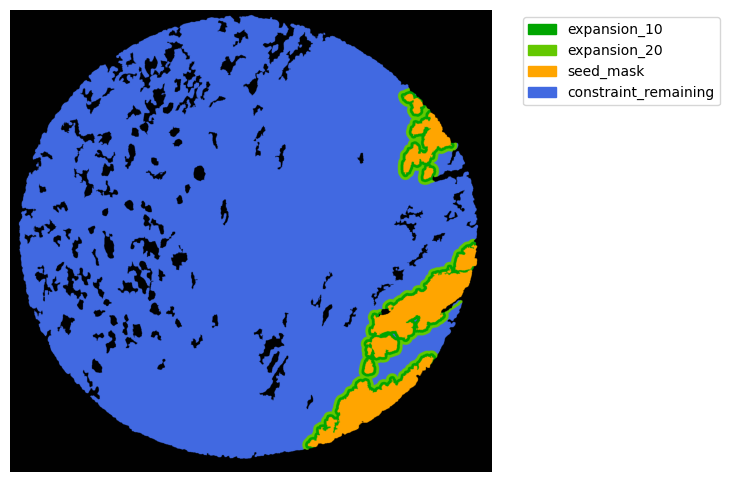
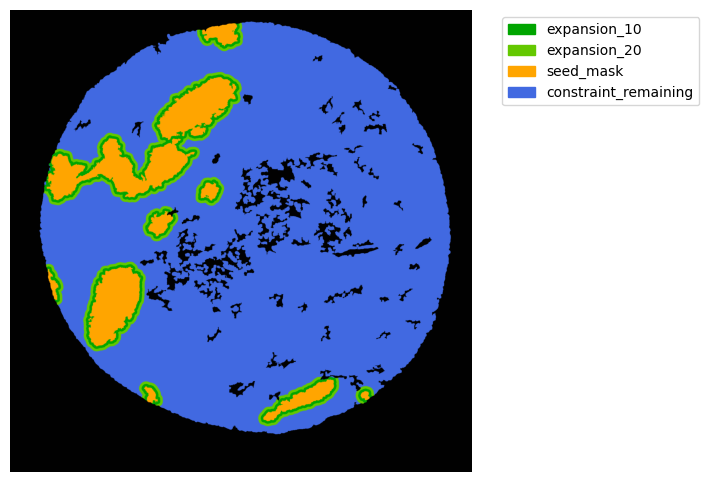
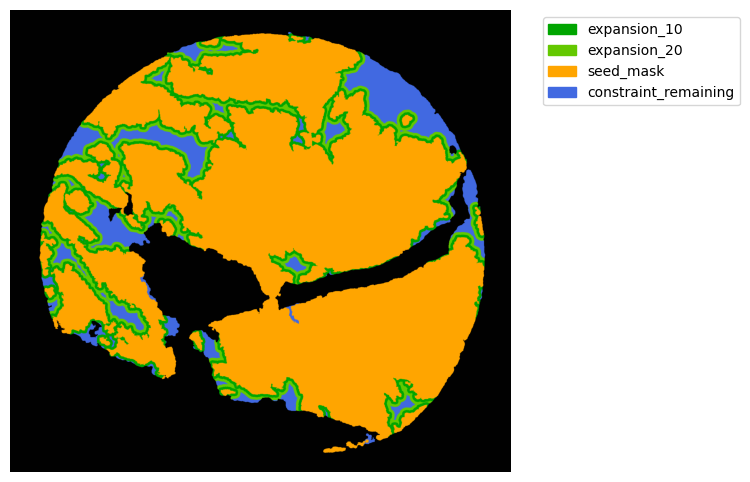
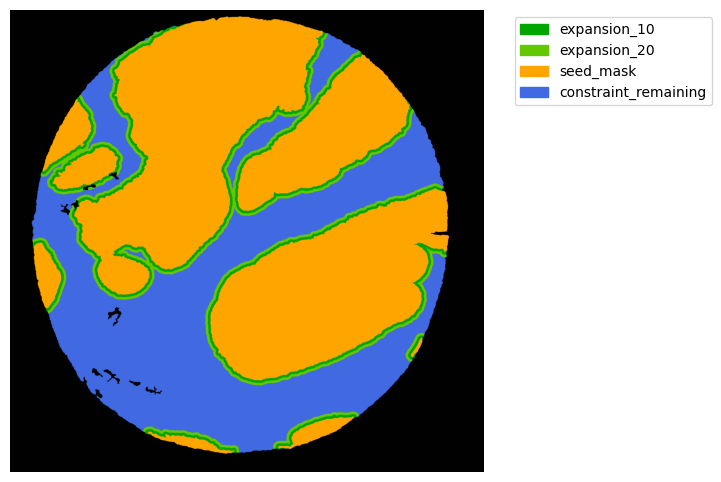
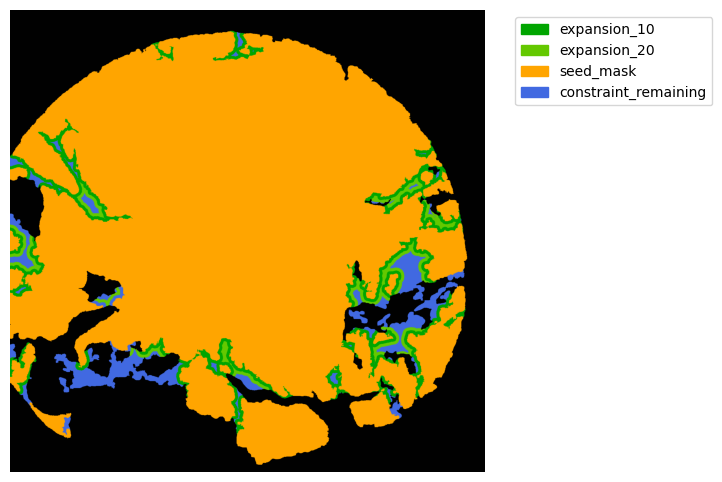
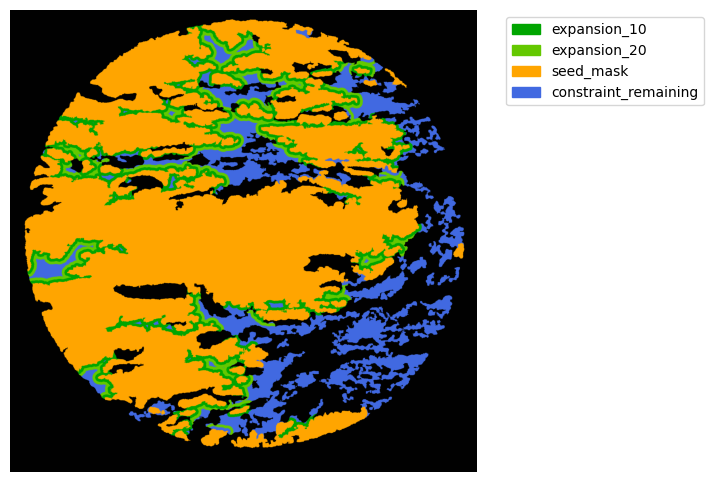
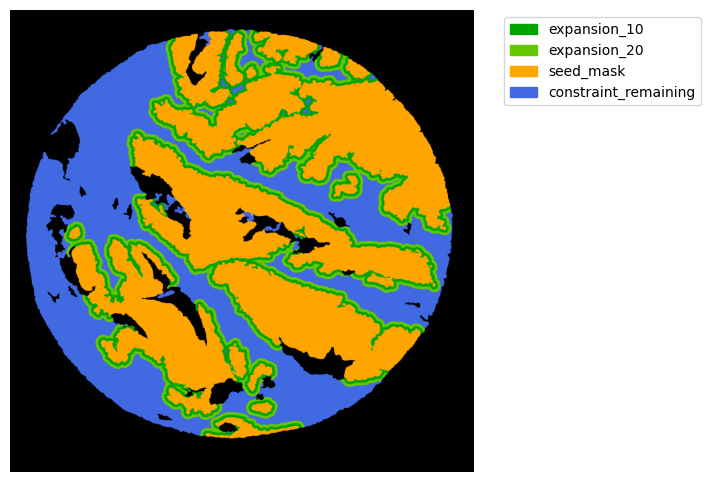
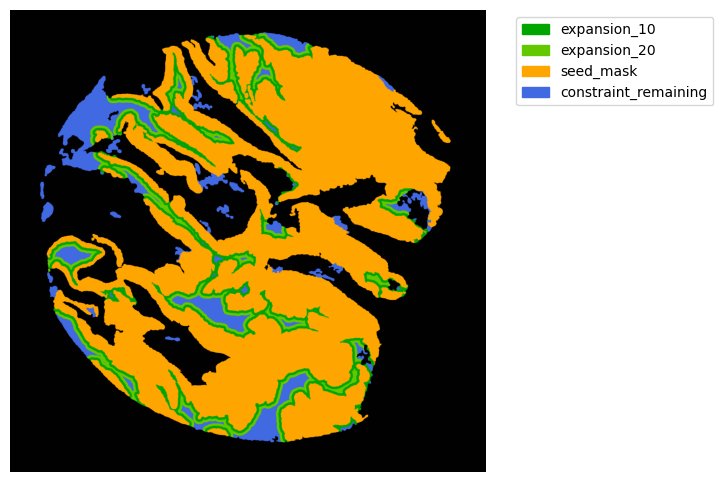
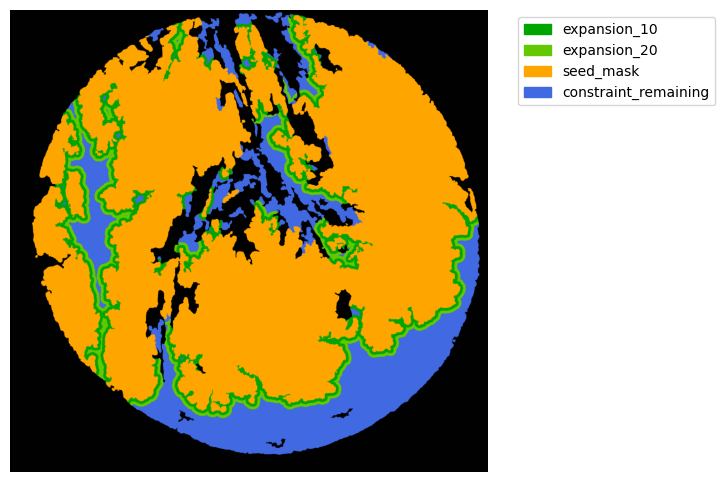
Now with cancer and stroma masks we can derive the interface.
The interface is the Cancer region expanded inside the stroma area. We will expand 10 and 20 um.
%autoreload
from gridgene import get_arrays as ga
from gridgene import contours, get_masks
# from GRIDGEN.src.mask_properties2 import GetMasksProperties
from gridgene.mask_properties2 import MaskAnalysisPipeline
TA = get_masks.ConstrainedMaskExpansion(mask_tum, mask_stroma)
TA.expand_mask(expansion_pixels=[10,20], min_area=1000)
# change names and colos«rs!
mask_colors = {'expansion_10':(0, 165, 0),
'expansion_20':(100, 200, 0),
'seed_mask':(255, 165, 0),
'constraint_remaining':(65, 105, 225) } # Strom}
GM.plot_masks(masks=TA.binary_expansions.values(),
mask_names=TA.binary_expansions.keys(),
background_color=(1, 1, 1), mask_colors=mask_colors, path=None, show=True, ax=None,
figsize=(8, 6))
2025-06-13 15:17:01,377 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Finally we can extract the information from the masks
# 1. Define your masks using MaskDefinition
mask_definitions = [
MaskDefinition(mask=TA.binary_expansions['constraint_remaining'], mask_name='Stroma_remaining', analysis_type='bulk'),
MaskDefinition(mask=TA.binary_expansions['seed_mask'], mask_name='Cancer', analysis_type='bulk'),
MaskDefinition(mask=TA.binary_expansions['expansion_10'], mask_name='CI_10um', analysis_type='bulk'),
MaskDefinition(mask=TA.binary_expansions['expansion_20'], mask_name='CI_20um', analysis_type='bulk'),
]
# 2. Initialize and run the analysis pipeline
pipeline = MaskAnalysisPipeline(mask_definitions=mask_definitions,
array_counts = array_total,
target_dict = target_dict_total,
)
results = pipeline.run()
df = pipeline.get_results_df()
df
run took 8.3321 seconds
| area | object_id | IL6ST | IGHG1 | COL1A1 | CTSH | SIRPA | TIMP2 | SPARC | LMO2 | ... | IFNB1 | IGHD | CD45RA | CCL19 | CD7 | EBI3 | LST1 | VPREB3 | mask_name | analysis_type | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1749628 | bulk | 5880 | 2682 | 20626 | 1271 | 1022 | 13634 | 31112 | 461 | ... | 40 | 6 | 3 | 40 | 14 | 26 | 0 | 8 | Stroma_remaining | bulk |
| 1 | 217909 | bulk | 1304 | 212 | 890 | 2249 | 758 | 754 | 1281 | 71 | ... | 23 | 6 | 0 | 17 | 10 | 11 | 1 | 4 | Cancer | bulk |
| 2 | 31943 | bulk | 163 | 60 | 910 | 174 | 97 | 399 | 1212 | 25 | ... | 1 | 0 | 0 | 3 | 2 | 7 | 0 | 0 | CI_10um | bulk |
| 3 | 31051 | bulk | 163 | 127 | 793 | 93 | 45 | 380 | 1049 | 22 | ... | 1 | 1 | 0 | 2 | 0 | 12 | 0 | 0 | CI_20um | bulk |
4 rows × 484 columns
We will now set up the code and apply the Cancer Stroma interface analysis to the cohort. We will save the results.
start_time = time.time()
save_path_ = 'results/tb_xenium_gr_/'
if not os.path.exists(save_path_):
os.makedirs(save_path_)
logger = logging.getLogger('contour_logger')
handler = logging.FileHandler(f'{save_path_}/log_file.log') # Change this to your desired log file path
formatter = logging.Formatter('%(asctime)s - %(name)s - %(levelname)s - %(message)s')
handler.setFormatter(formatter)
logger.addHandler(handler)
logger.setLevel(logging.INFO)
logger.propagate = False
# TUM / STROMA + TM BORDER
for file_csv in tqdm(files, desc="Processing Folders", unit="folder"):
start_time_lap = time.time()
logger.info(f'Processing folder {file_csv}')
base_name = os.path.basename(file_csv)
file_name = "_".join(base_name.split("_")[:2])
save_path = os.path.join(save_path_, file_name)
if not os.path.exists(save_path):
os.makedirs(save_path)
df_total = pd.read_csv(file_csv)
df_total = df_total[['x_location', 'y_location', 'feature_name']]
df_total = df_total.rename(columns={'feature_name': 'target'})
df_total = df_total[~df_total['target'].str.contains('System|egative')]
df_total['X'] = df_total['x_location'] - min(df_total['x_location'])
df_total['Y'] = df_total['y_location'] - min(df_total['y_location'])
n_genes = len(df_total['target'].unique())
height = int(max(df_total['X'])) + 1
width = int(max(df_total['Y'])) + 1
# define arrays
target_dict_total = {target: index for index, target in enumerate(df_total['target'].unique())}
array_total = ga.transform_df_to_array(df = df_total, target_dict=target_dict_total, array_shape = (height, width,len(target_dict_total))).astype(np.int8)
df_subset_tum, array_subset_tum, target_indices_subset_tum = ga.get_subset_arrays(df_total, array_total,target_dict_total,
target_list=target_tum, target_col = 'target')
# obtain contours
CTum = contours.ConvolutionContours(array_subset_tum, contour_name='tum')
CTum.get_conv_sum(kernel_size=kernel_size_tum, kernel_shape='square')
CTum.contours_from_sum(density_threshold = density_th_tum,
min_area_threshold = min_area_th_tum , directionality = 'higher')
CEmpty = contours.ConvolutionContours(array_total, contour_name='empty')
CEmpty.get_conv_sum(kernel_size=kernel_size_empty, kernel_shape='square')
CEmpty.contours_from_sum(density_threshold = density_th_empty,
min_area_threshold = min_area_th_empty, directionality = 'lower') # attention that directionality is lower here
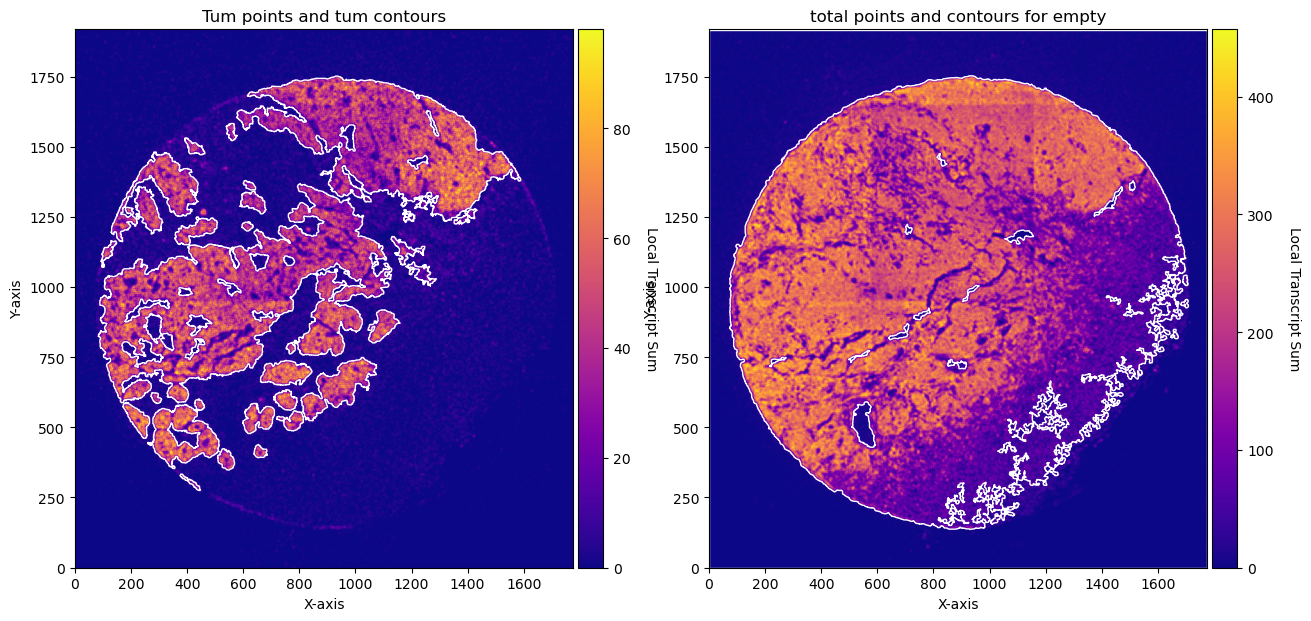
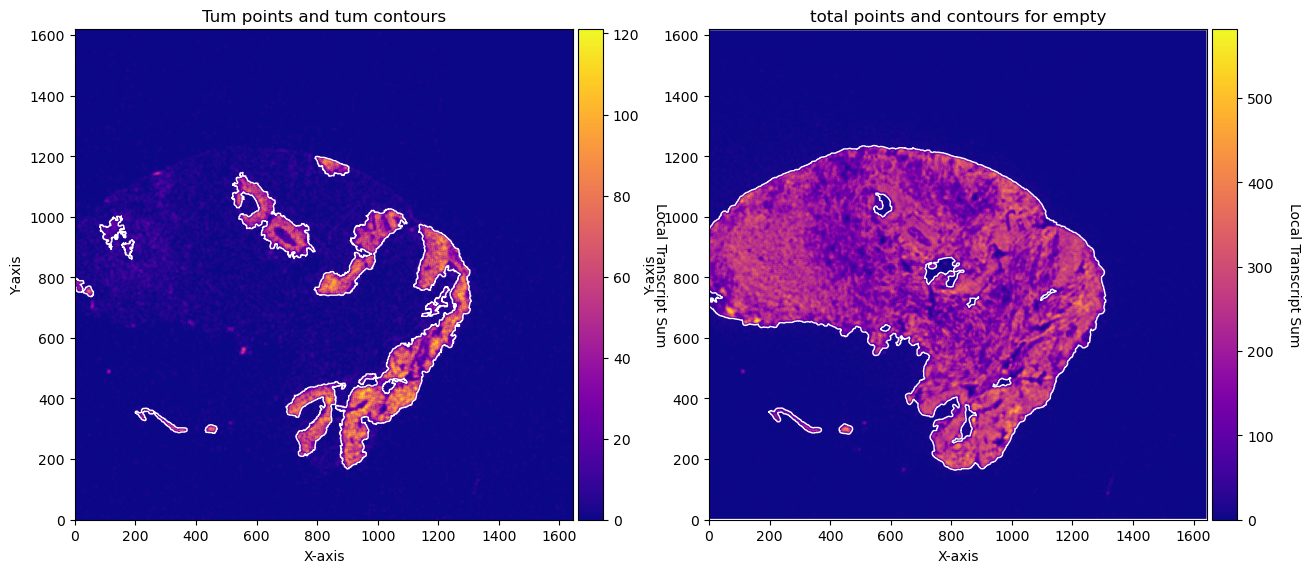
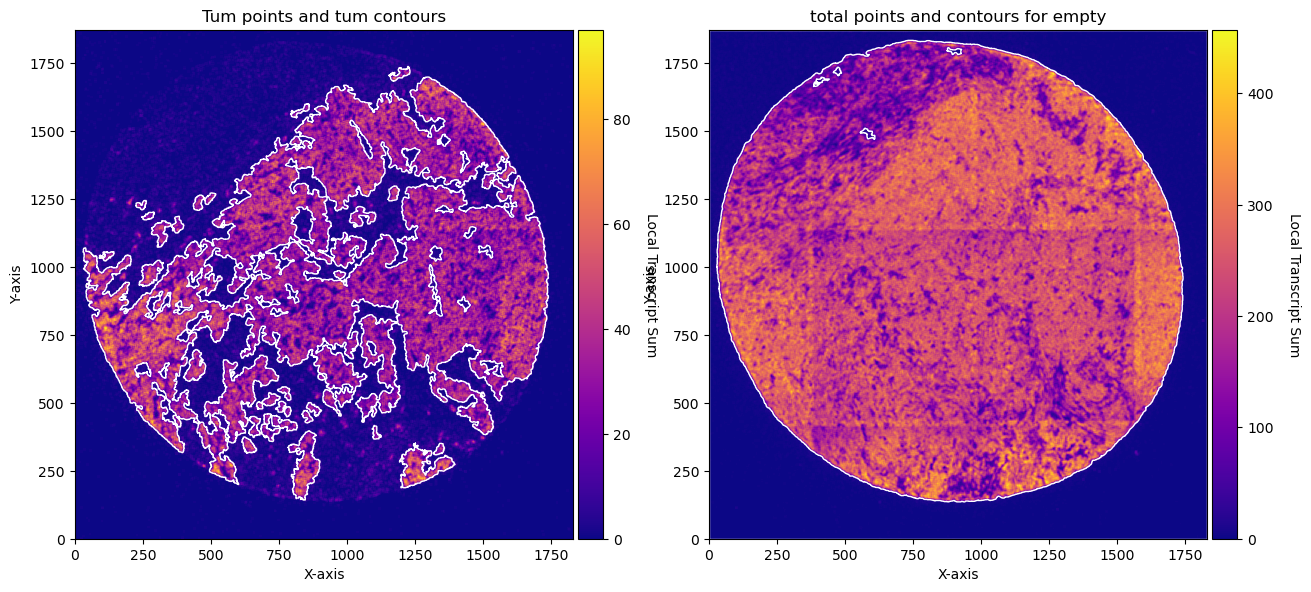
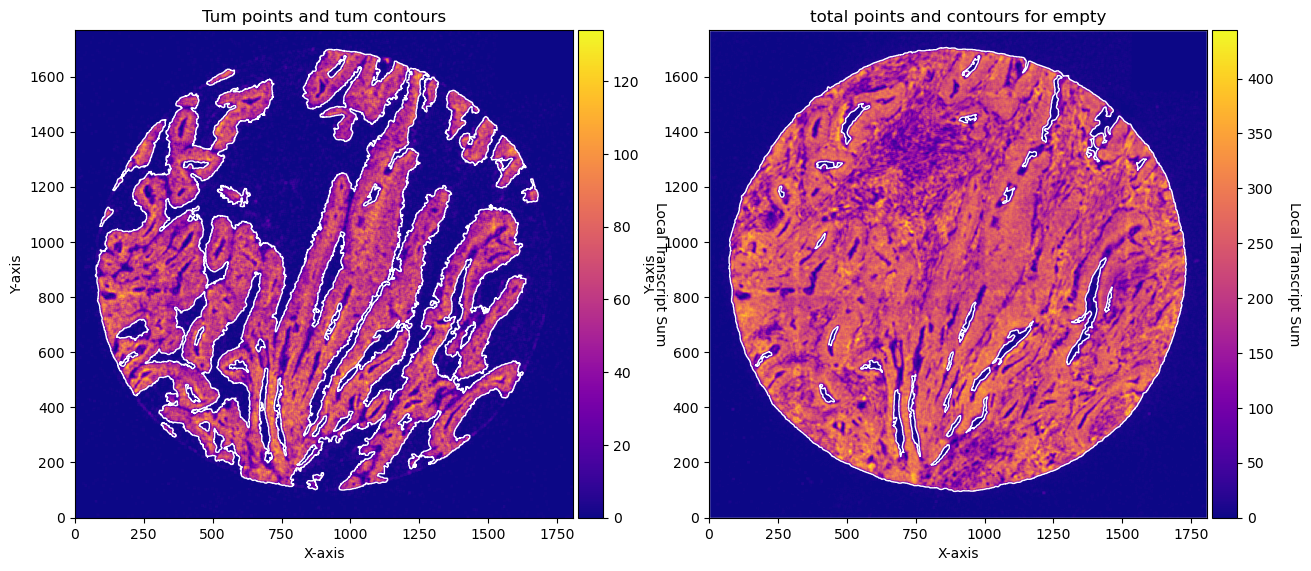
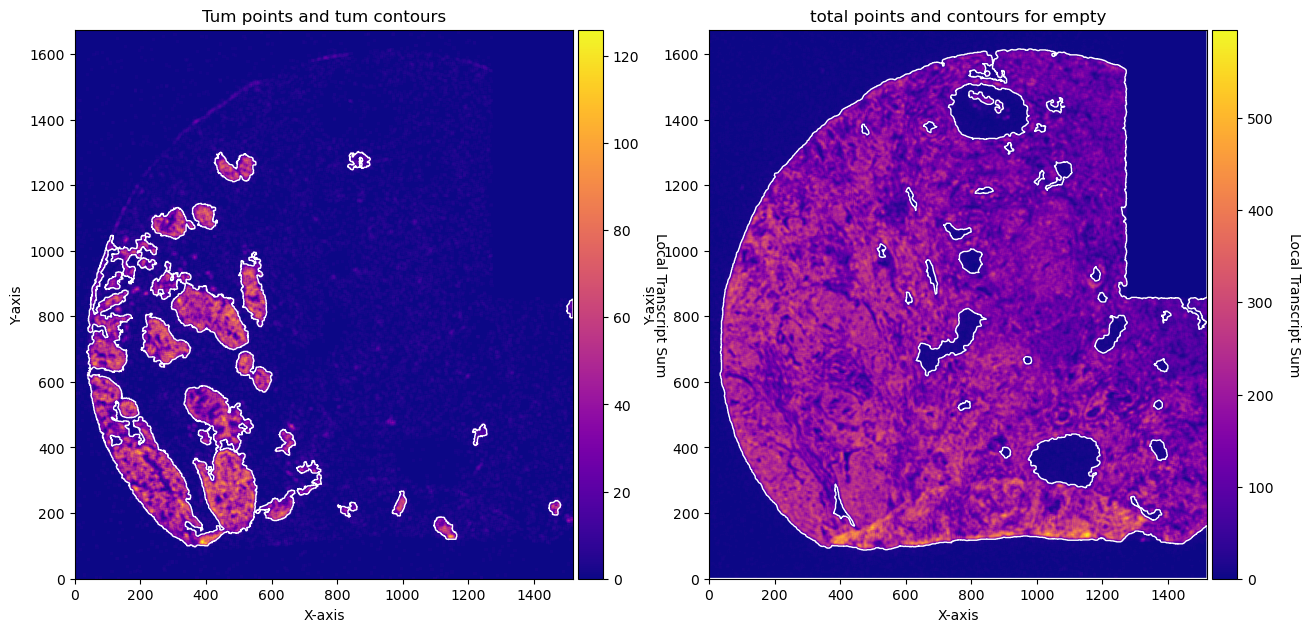
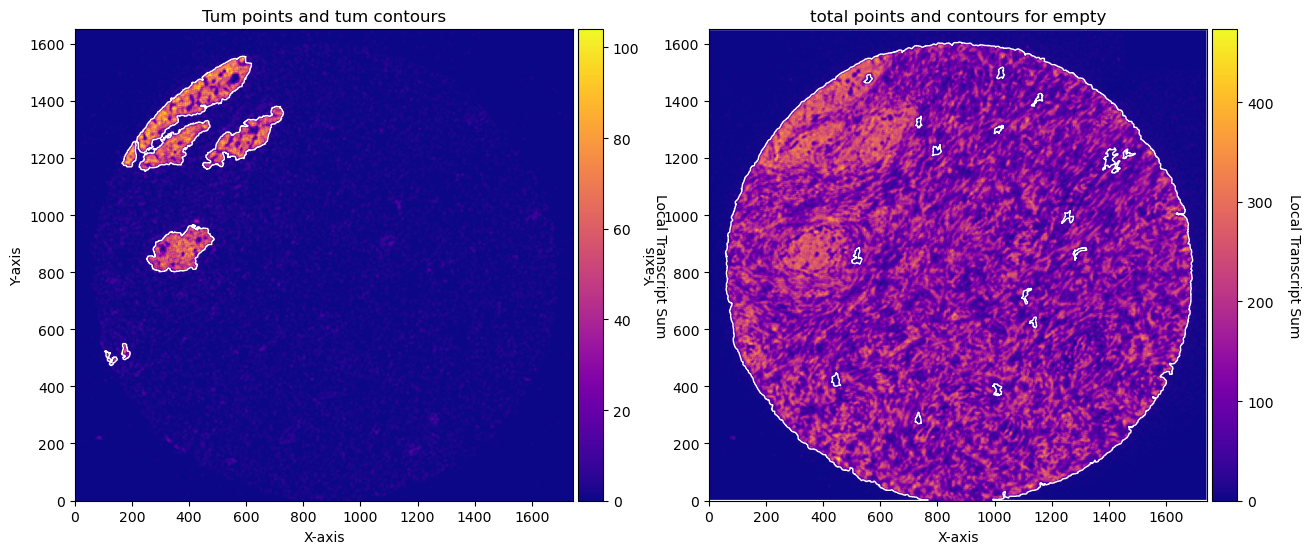
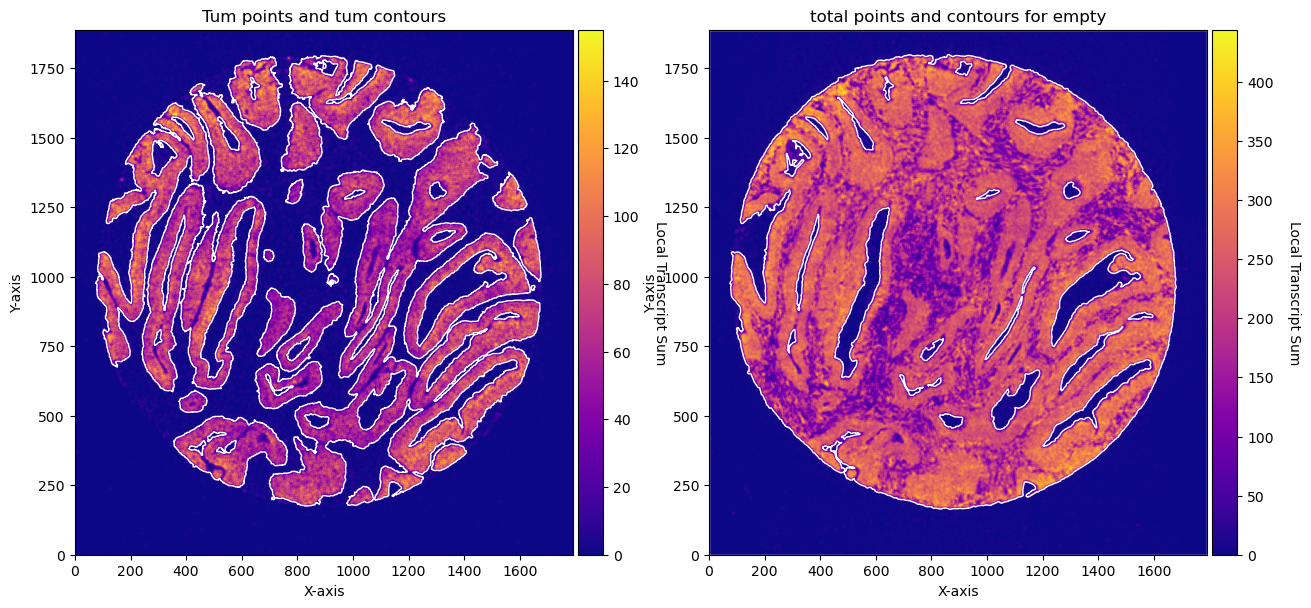
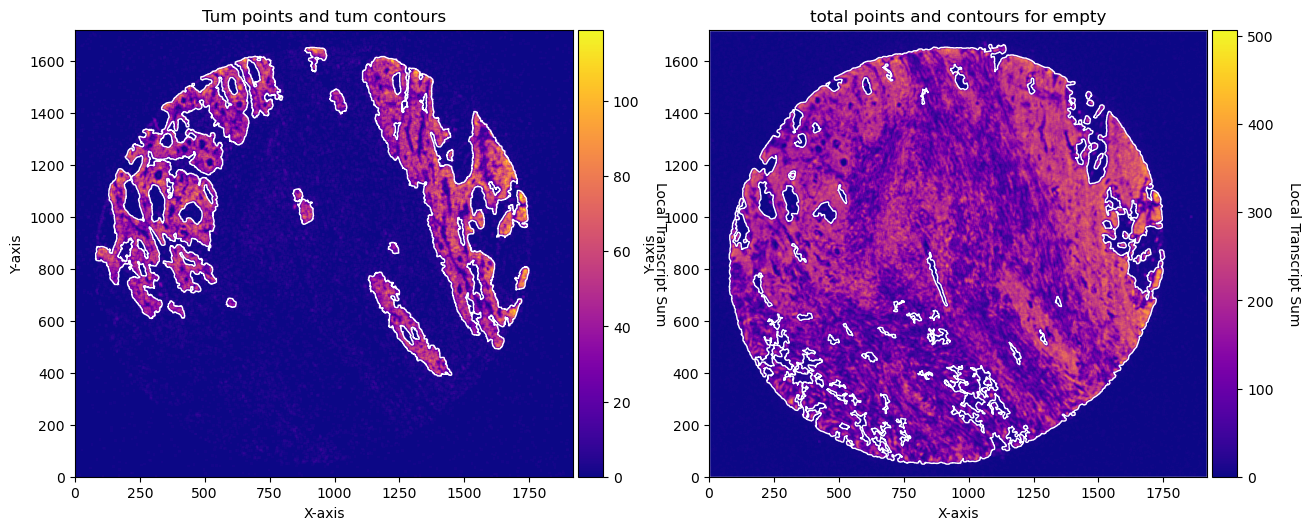
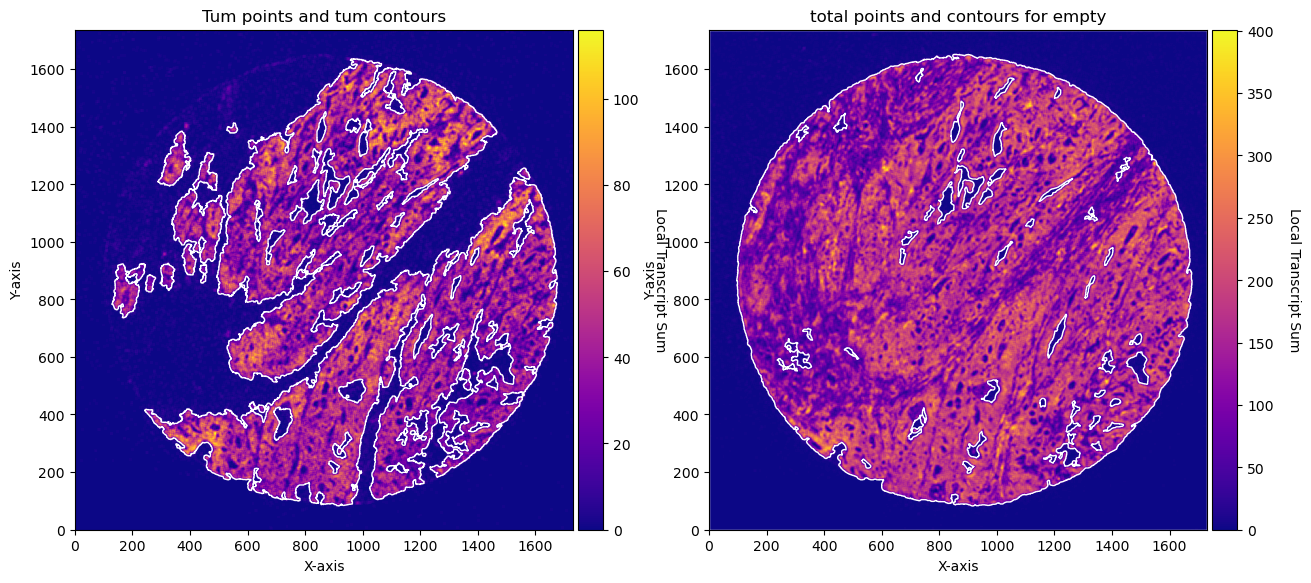
fig, axs = plt.subplots(1, 2, figsize=(15, 10))
CTum.plot_conv_sum(cmap='plasma', c_countour='white', ax=axs[0])
axs[0].set_title('Tum points and tum contours')
CEmpty.plot_conv_sum(cmap='plasma', c_countour='white', ax=axs[1])
axs[1].set_title('total points and contours for empty')
plt.show()
#### obtain masks
GM = get_masks.GetMasks(image_shape = (height, width))
mask_empty = GM.create_mask(CEmpty.contours)
mask_tum = GM.create_mask(CTum.contours)
mask_tum = GM.fill_holes(mask_tum)
mask_stroma = GM.subtract_masks(np.ones((height, width), dtype=np.uint8), mask_tum, mask_empty)
mask_stroma = GM.filter_binary_mask_by_area(mask_stroma, min_area=700)
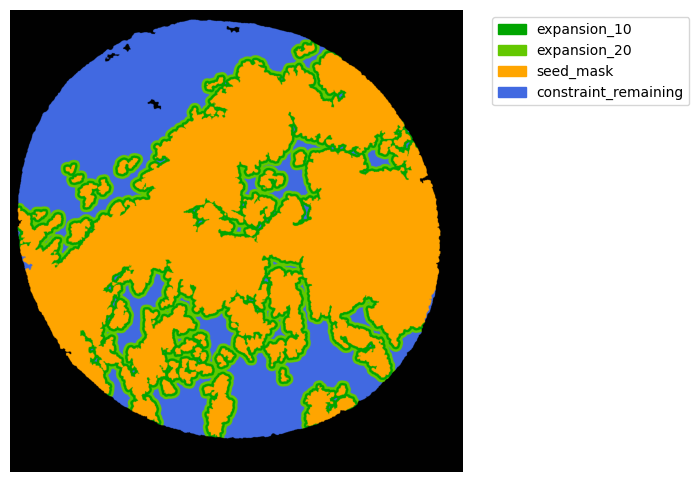
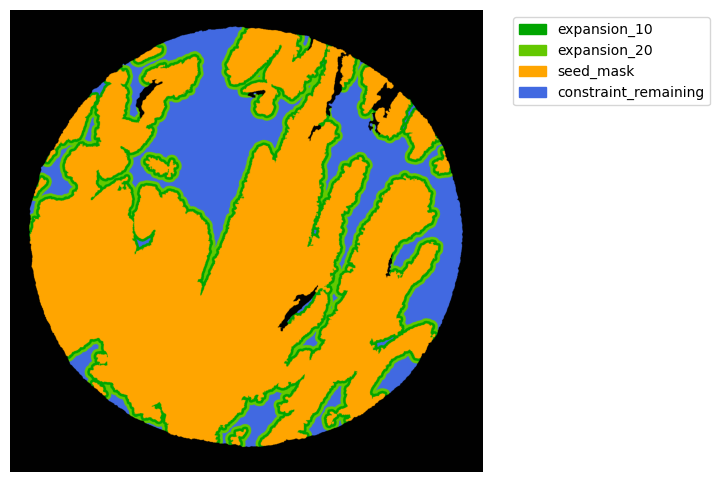
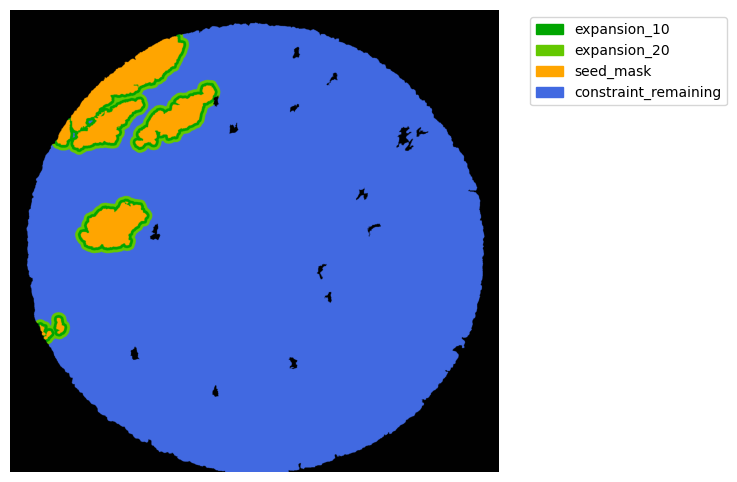
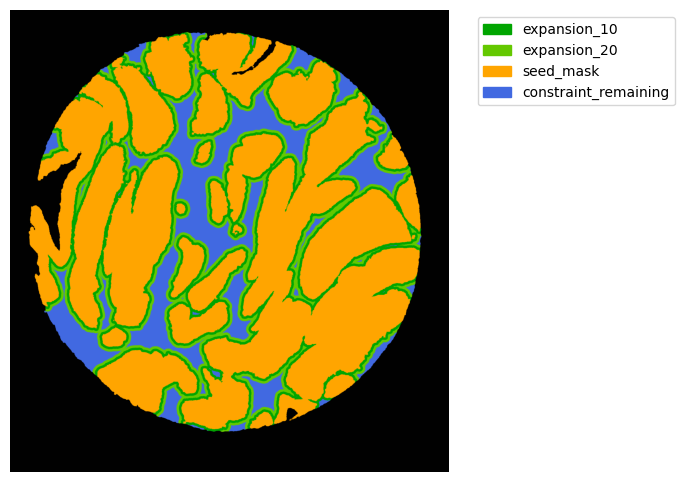
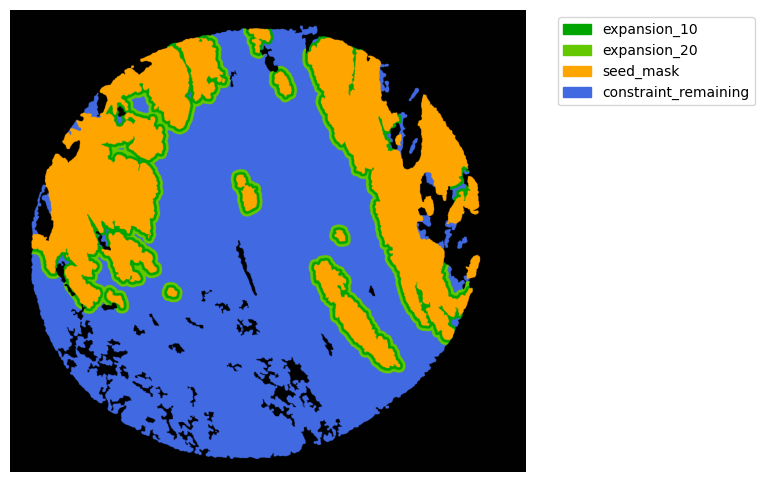
#### Interface analysis
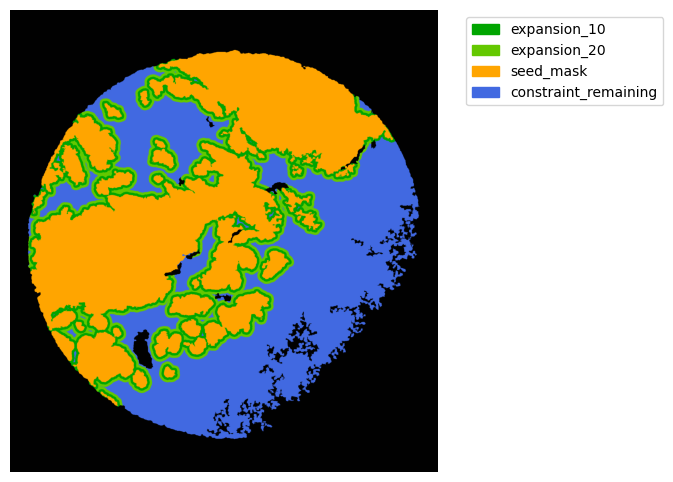
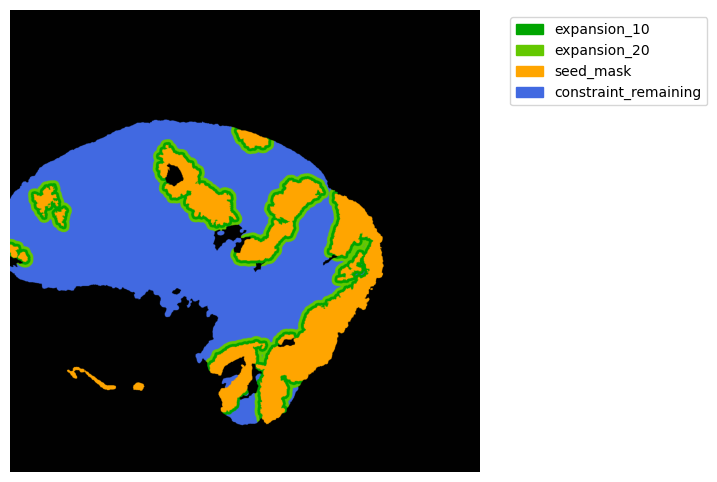
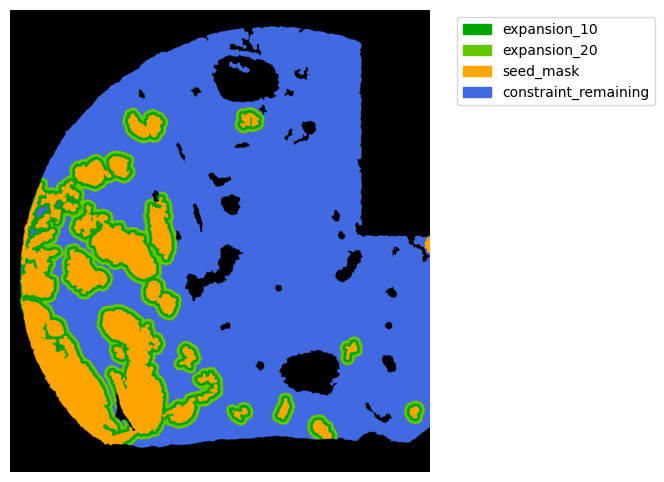
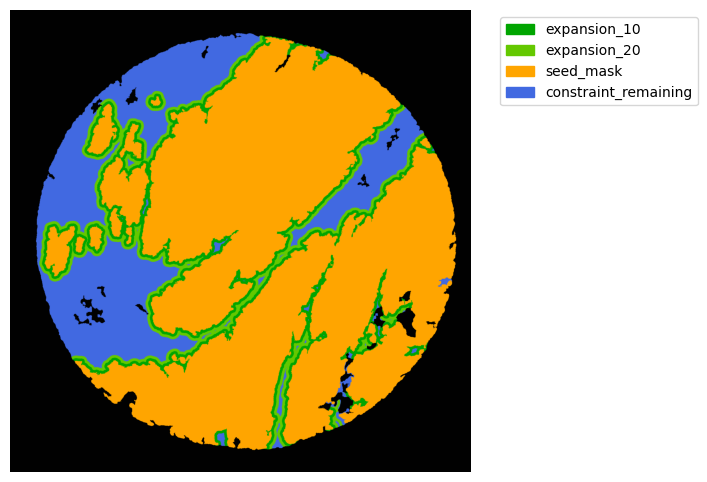
TA = get_masks.ConstrainedMaskExpansion(mask_tum, mask_stroma)
TA.expand_mask(expansion_pixels=[10,20], min_area=1000)
# change names and colos«rs!
mask_colors = {'expansion_10':(0, 165, 0),
'expansion_20':(100, 200, 0),
'seed_mask':(255, 165, 0),
'constraint_remaining':(65, 105, 225) } # Strom}
GM.plot_masks(masks=TA.binary_expansions.values(),
mask_names=TA.binary_expansions.keys(),
background_color=(1, 1, 1), mask_colors=mask_colors, path=None, show=True, ax=None,
figsize=(8, 6))
mask_definitions = [
MaskDefinition(mask=TA.binary_expansions['constraint_remaining'], mask_name='Stroma_remaining', analysis_type='per_object'),
MaskDefinition(mask=TA.binary_expansions['seed_mask'], mask_name='Cancer', analysis_type='per_object'),
MaskDefinition(mask=TA.binary_expansions['expansion_10'], mask_name='CI_10um', analysis_type='per_object'),
MaskDefinition(mask=TA.binary_expansions['expansion_20'], mask_name='CI_20um', analysis_type='per_object'),
]
# 2. Initialize and run the analysis pipeline
pipeline = MaskAnalysisPipeline(mask_definitions=mask_definitions,
array_counts = array_total,
target_dict = target_dict_total,
)
results = pipeline.run()
df = pipeline.get_results_df()
df.to_csv(os.path.join(save_path + f'/{file_name}_tb.csv'), index=False)
# Record the end time
end_time = time.time()
# Calculate the duration
duration = start_time - time.time()
logging.info(f'full analysis in {duration}seconds')
Processing Folders: 0%| | 0/20 [00:00<?, ?folder/s]2025-06-13 15:24:48,699 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection23_filtered.csv
2025-06-13 15:24:53,483 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:24:53,628 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 29
2025-06-13 15:24:53,629 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0813 seconds
contours_from_sum took 0.0635 seconds
get_conv_sum took 0.6328 seconds
2025-06-13 15:24:54,905 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 94
contours_from_sum took 0.6411 seconds
2025-06-13 15:24:55,597 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:24:55,617 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:24:55,736 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 5%|██████ | 1/20 [00:16<05:09, 16.30s/folder]2025-06-13 15:25:04,995 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection21_filtered.csv
run took 8.5054 seconds
2025-06-13 15:25:10,567 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:25:10,727 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 43
2025-06-13 15:25:10,728 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0830 seconds
contours_from_sum took 0.0766 seconds
get_conv_sum took 0.7076 seconds
2025-06-13 15:25:12,086 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 40
contours_from_sum took 0.6504 seconds
2025-06-13 15:25:12,737 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:25:12,755 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:25:12,824 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 10%|████████████ | 2/20 [00:33<05:01, 16.75s/folder]2025-06-13 15:25:22,069 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection17_filtered.csv
run took 8.6011 seconds
2025-06-13 15:25:25,416 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:25:25,543 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 10
2025-06-13 15:25:25,544 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0696 seconds
contours_from_sum took 0.0576 seconds
get_conv_sum took 0.5580 seconds
2025-06-13 15:25:26,665 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 114
contours_from_sum took 0.5631 seconds
2025-06-13 15:25:27,284 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:25:27,299 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:25:27,316 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 15%|██████████████████ | 3/20 [00:46<04:14, 14.99s/folder]2025-06-13 15:25:34,956 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection31_filtered.csv
run took 7.1751 seconds
2025-06-13 15:25:37,672 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:25:37,796 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 14
2025-06-13 15:25:37,796 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0654 seconds
contours_from_sum took 0.0584 seconds
get_conv_sum took 0.5543 seconds
2025-06-13 15:25:38,894 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 11
contours_from_sum took 0.5432 seconds
2025-06-13 15:25:39,433 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:25:39,447 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:25:39,474 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 20%|████████████████████████ | 4/20 [00:58<03:40, 13.76s/folder]2025-06-13 15:25:46,821 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection7_filtered.csv
run took 6.8109 seconds
2025-06-13 15:25:50,694 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:25:50,831 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 10
2025-06-13 15:25:50,832 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0812 seconds
contours_from_sum took 0.0559 seconds
get_conv_sum took 0.6345 seconds
2025-06-13 15:25:52,047 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 78
contours_from_sum took 0.5817 seconds
2025-06-13 15:25:52,676 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:25:52,691 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:25:52,714 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 25%|██████████████████████████████ | 5/20 [01:13<03:32, 14.16s/folder]2025-06-13 15:26:01,706 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection11_filtered.csv
run took 8.4426 seconds
2025-06-13 15:26:09,129 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:26:09,298 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 55
2025-06-13 15:26:09,298 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0832 seconds
contours_from_sum took 0.0849 seconds
get_conv_sum took 0.6679 seconds
2025-06-13 15:26:10,607 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 6
contours_from_sum took 0.6403 seconds
2025-06-13 15:26:11,363 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:26:11,377 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:26:11,422 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 30%|████████████████████████████████████ | 6/20 [01:32<03:41, 15.85s/folder]2025-06-13 15:26:20,845 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection25_filtered.csv
run took 8.7425 seconds
2025-06-13 15:26:27,196 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:26:27,360 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 36
2025-06-13 15:26:27,360 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0827 seconds
contours_from_sum took 0.0808 seconds
get_conv_sum took 0.7205 seconds
2025-06-13 15:26:28,787 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 8
contours_from_sum took 0.7060 seconds
2025-06-13 15:26:29,418 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:26:29,436 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:26:29,496 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 35%|██████████████████████████████████████████ | 7/20 [01:50<03:35, 16.55s/folder]2025-06-13 15:26:38,816 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection22_filtered.csv
run took 8.6170 seconds
2025-06-13 15:26:45,233 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:26:45,392 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 40
2025-06-13 15:26:45,393 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0814 seconds
contours_from_sum took 0.0776 seconds
get_conv_sum took 0.6747 seconds
2025-06-13 15:26:46,732 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 29
contours_from_sum took 0.6644 seconds
2025-06-13 15:26:47,368 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:26:47,386 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:26:47,435 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 40%|████████████████████████████████████████████████ | 8/20 [02:07<03:22, 16.90s/folder]2025-06-13 15:26:56,472 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection30_filtered.csv
run took 8.1412 seconds
2025-06-13 15:27:02,064 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:27:02,201 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 15
2025-06-13 15:27:02,202 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0789 seconds
contours_from_sum took 0.0582 seconds
get_conv_sum took 0.5776 seconds
2025-06-13 15:27:03,358 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 17
contours_from_sum took 0.5787 seconds
2025-06-13 15:27:04,007 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:27:04,021 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:27:04,093 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 45%|██████████████████████████████████████████████████████ | 9/20 [02:24<03:05, 16.84s/folder]2025-06-13 15:27:13,193 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection20_filtered.csv
run took 8.4403 seconds
2025-06-13 15:27:16,320 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:27:16,463 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 7
2025-06-13 15:27:16,464 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0802 seconds
contours_from_sum took 0.0626 seconds
get_conv_sum took 0.5847 seconds
2025-06-13 15:27:17,734 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 29
contours_from_sum took 0.6858 seconds
2025-06-13 15:27:18,346 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:27:18,358 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:27:18,409 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 50%|███████████████████████████████████████████████████████████▌ | 10/20 [02:38<02:39, 15.98s/folder]2025-06-13 15:27:27,249 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection16_filtered.csv
run took 8.2607 seconds
2025-06-13 15:27:31,353 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:27:31,472 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 25
2025-06-13 15:27:31,472 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0625 seconds
contours_from_sum took 0.0555 seconds
get_conv_sum took 0.5592 seconds
2025-06-13 15:27:32,571 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 33
contours_from_sum took 0.5396 seconds
2025-06-13 15:27:33,141 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:27:33,156 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:27:33,176 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 55%|█████████████████████████████████████████████████████████████████▍ | 11/20 [02:51<02:15, 15.04s/folder]2025-06-13 15:27:40,140 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection26_filtered.csv
run took 6.3943 seconds
2025-06-13 15:27:45,103 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:27:45,210 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 42
2025-06-13 15:27:45,211 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0580 seconds
contours_from_sum took 0.0492 seconds
get_conv_sum took 0.4936 seconds
2025-06-13 15:27:46,195 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 42
contours_from_sum took 0.4902 seconds
2025-06-13 15:27:46,780 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:27:46,797 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:27:46,877 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 60%|███████████████████████████████████████████████████████████████████████▍ | 12/20 [03:04<01:56, 14.58s/folder]2025-06-13 15:27:53,662 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection8_filtered.csv
run took 6.1695 seconds
2025-06-13 15:27:57,985 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:27:58,107 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 7
2025-06-13 15:27:58,107 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0714 seconds
contours_from_sum took 0.0502 seconds
get_conv_sum took 0.5146 seconds
2025-06-13 15:27:59,129 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 17
contours_from_sum took 0.5074 seconds
2025-06-13 15:27:59,723 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:27:59,737 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:27:59,754 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 65%|█████████████████████████████████████████████████████████████████████████████▎ | 13/20 [03:18<01:40, 14.40s/folder]2025-06-13 15:28:07,657 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection29_filtered.csv
run took 7.4426 seconds
2025-06-13 15:28:10,960 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:28:11,104 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 68
2025-06-13 15:28:11,105 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0685 seconds
contours_from_sum took 0.0752 seconds
get_conv_sum took 0.4979 seconds
2025-06-13 15:28:12,102 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 120
contours_from_sum took 0.4993 seconds
2025-06-13 15:28:12,774 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:28:12,796 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:28:12,896 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 70%|███████████████████████████████████████████████████████████████████████████████████▎ | 14/20 [03:32<01:24, 14.02s/folder]2025-06-13 15:28:20,809 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection13_filtered.csv
run took 7.0631 seconds
2025-06-13 15:28:26,650 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:28:26,805 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 50
2025-06-13 15:28:26,805 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0826 seconds
contours_from_sum took 0.0718 seconds
get_conv_sum took 0.7140 seconds
2025-06-13 15:28:28,206 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 28
contours_from_sum took 0.6869 seconds
2025-06-13 15:28:28,885 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:28:28,903 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:28:28,981 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 75%|█████████████████████████████████████████████████████████████████████████████████████████▎ | 15/20 [03:49<01:15, 15.08s/folder]2025-06-13 15:28:38,339 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection12_filtered.csv
run took 8.6615 seconds
2025-06-13 15:28:44,303 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:28:44,441 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 25
2025-06-13 15:28:44,442 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0769 seconds
contours_from_sum took 0.0616 seconds
get_conv_sum took 0.6044 seconds
2025-06-13 15:28:45,575 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 39
contours_from_sum took 0.5282 seconds
2025-06-13 15:28:46,181 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:28:46,189 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:28:46,225 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 80%|███████████████████████████████████████████████████████████████████████████████████████████████▏ | 16/20 [04:06<01:01, 15.48s/folder]2025-06-13 15:28:54,761 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection28_filtered.csv
run took 7.7920 seconds
2025-06-13 15:28:59,173 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:28:59,310 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 36
2025-06-13 15:28:59,310 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0769 seconds
contours_from_sum took 0.0598 seconds
get_conv_sum took 0.6886 seconds
2025-06-13 15:29:00,574 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 67
contours_from_sum took 0.5752 seconds
2025-06-13 15:29:01,208 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:29:01,214 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:29:01,260 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 85%|█████████████████████████████████████████████████████████████████████████████████████████████████████▏ | 17/20 [04:21<00:46, 15.45s/folder]2025-06-13 15:29:10,145 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection34_filtered.csv
run took 8.2578 seconds
2025-06-13 15:29:14,999 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:29:15,155 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 28
2025-06-13 15:29:15,155 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0866 seconds
contours_from_sum took 0.0686 seconds
get_conv_sum took 0.7575 seconds
2025-06-13 15:29:16,650 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 52
contours_from_sum took 0.7374 seconds
2025-06-13 15:29:17,430 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:29:17,450 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:29:17,535 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 90%|███████████████████████████████████████████████████████████████████████████████████████████████████████████ | 18/20 [04:38<00:32, 16.02s/folder]2025-06-13 15:29:27,479 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection9_filtered.csv
run took 9.2704 seconds
/tmp/ipykernel_793845/3712573084.py:24: DtypeWarning: Columns (10) have mixed types. Specify dtype option on import or set low_memory=False.
df_total = pd.read_csv(file_csv)
2025-06-13 15:29:31,787 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:29:31,928 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 56
2025-06-13 15:29:31,928 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0750 seconds
contours_from_sum took 0.0645 seconds
get_conv_sum took 0.5177 seconds
2025-06-13 15:29:32,963 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 48
contours_from_sum took 0.5166 seconds
2025-06-13 15:29:33,612 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:29:33,629 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:29:33,673 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 95%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████ | 19/20 [04:53<00:15, 15.54s/folder]2025-06-13 15:29:41,890 - contour_logger - INFO - Processing folder ../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection19_filtered.csv
run took 7.6432 seconds
2025-06-13 15:29:45,775 - gridgen.contours.tum - INFO - Initialized GetContour
2025-06-13 15:29:45,909 - gridgen.contours.tum - INFO - Number of contours after filtering no counts: 38
2025-06-13 15:29:45,909 - gridgen.contours.empty - INFO - Initialized GetContour
get_conv_sum took 0.0672 seconds
contours_from_sum took 0.0664 seconds
get_conv_sum took 0.5556 seconds
2025-06-13 15:29:47,016 - gridgen.contours.empty - INFO - Number of contours after filtering no counts: 61
contours_from_sum took 0.5510 seconds
2025-06-13 15:29:47,631 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
2025-06-13 15:29:47,639 - gridgen.get_masks.GetMasks - INFO - Subtracted masks from base mask.
2025-06-13 15:29:47,696 - gridgen.get_masks.GetMasks - INFO - Initialized GetMasks
Processing Folders: 100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 20/20 [05:06<00:00, 15.34s/folder]
run took 7.2035 seconds
Analysis of the results from cancer stroma interface
To be noted that each total mask per sample is one object. This is, per sample, we have one cancer object, one stroma object, one 10 um interface and one 20 um interface.
Grab information stored in csv
save_path_ = 'results/tb_xenium_gr_/'
dfs = []
for file in files:
base_name = os.path.basename(file)
selection_name = "_".join(base_name.split("_")[:2])
df = pd.read_csv(f'{save_path_}/{selection_name}/{selection_name}_tb.csv')
df['selection'] = selection_name
dfs.append(df)
full_df = pd.concat(dfs, ignore_index=True)
full_df.head()
| area | object_id | BTN2A2 | NT5E | MDM2 | LGALS3 | EMP1 | PODXL | SPARC | REG1A | ... | EBI3 | CXCL14 | CSF2 | TNFRSF18 | LST1 | VPREB3 | CD45RA | mask_name | analysis_type | selection | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 335788 | bulk | 136 | 362 | 642 | 649 | 2303 | 597 | 12760 | 2600 | ... | 16 | 31 | 7 | 4 | 1 | 0 | 0.0 | Stroma_remaining | bulk | TMA1_Selection23 |
| 1 | 1265974 | bulk | 1260 | 12727 | 9217 | 34220 | 7574 | 4859 | 17094 | 278407 | ... | 27 | 287 | 39 | 7 | 4 | 3 | 2.0 | Cancer | bulk | TMA1_Selection23 |
| 2 | 169633 | bulk | 77 | 295 | 390 | 471 | 1948 | 689 | 13848 | 5154 | ... | 14 | 45 | 2 | 1 | 0 | 0 | 0.0 | CI_10um | bulk | TMA1_Selection23 |
| 3 | 88214 | bulk | 32 | 119 | 215 | 210 | 1037 | 308 | 7496 | 1188 | ... | 5 | 29 | 0 | 4 | 0 | 0 | 0.0 | CI_20um | bulk | TMA1_Selection23 |
| 4 | 723049 | bulk | 405 | 1654 | 1419 | 2402 | 7528 | 1021 | 26972 | 171 | ... | 53 | 74 | 480 | 8 | 9 | 9 | 0.0 | Stroma_remaining | bulk | TMA1_Selection21 |
5 rows × 485 columns
Transform into adata object
obs_columns = ['area', 'object_id', 'mask_name','analysis_type','selection',]
# 'perimeter', 'centroid', 'min_x', 'min_y', 'max_x', 'max_y', 'vertices',
# 'BoundingBox', 'mask_name', 'per_object', 'selection',]
counts_columns = full_df.columns.difference(obs_columns)
counts = full_df[counts_columns]
obs = full_df[obs_columns]
adata = ad.AnnData(counts)
adata.obs = obs
adata.layers["counts"] = adata.X.copy()
adata.obs = adata.obs.merge(metadata, left_on='selection', right_on='name', how='left')
#combine TB of 10 and 20in an unque TB
combined_replacements = {
'CI_10um': 'Interface',
'CI_20um': 'Interface',
'Stroma_remaining': 'Stroma'
}
obs['mask_name_combined'] = obs['mask_name'].replace(combined_replacements).astype('category')
adata.obs['mask_name_combined'] = obs['mask_name_combined']
print('number of cores: ', len(adata.obs['name'].unique()))
print('number of cores: ', len(adata.obs['name'].unique()))
adata.obs.head()
number of cores: 20
number of cores: 20
/home/martinha/miniconda3/envs/GRIDGEN/lib/python3.11/site-packages/anndata/utils.py:311: UserWarning: X converted to numpy array with dtype float64
warnings.warn(f"{name} converted to numpy array with dtype {arr.dtype}")
/home/martinha/miniconda3/envs/GRIDGEN/lib/python3.11/site-packages/anndata/_core/aligned_df.py:68: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
/tmp/ipykernel_793845/2097024825.py:23: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame.
Try using .loc[row_indexer,col_indexer] = value instead
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
obs['mask_name_combined'] = obs['mask_name'].replace(combined_replacements).astype('category')
| area | object_id | mask_name | analysis_type | selection | XENIUM_TMA | Selection | annotation | Hes | TMA | ... | Block | Unnamed: 8 | MMR | HLA | B2M abcam | IHC group | B2M gene defect | Final group | name | mask_name_combined | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 335788 | bulk | Stroma_remaining | bulk | TMA1_Selection23 | TMA1 | Selection 23 | NaN | yes | GD_TMA1 | ... | 1G | S00067 | MMRd | positive | positive | HLA+ | NaN | HLA+ | TMA1_Selection23 | Stroma |
| 1 | 1265974 | bulk | Cancer | bulk | TMA1_Selection23 | TMA1 | Selection 23 | NaN | yes | GD_TMA1 | ... | 1G | S00067 | MMRd | positive | positive | HLA+ | NaN | HLA+ | TMA1_Selection23 | Cancer |
| 2 | 169633 | bulk | CI_10um | bulk | TMA1_Selection23 | TMA1 | Selection 23 | NaN | yes | GD_TMA1 | ... | 1G | S00067 | MMRd | positive | positive | HLA+ | NaN | HLA+ | TMA1_Selection23 | Interface |
| 3 | 88214 | bulk | CI_20um | bulk | TMA1_Selection23 | TMA1 | Selection 23 | NaN | yes | GD_TMA1 | ... | 1G | S00067 | MMRd | positive | positive | HLA+ | NaN | HLA+ | TMA1_Selection23 | Interface |
| 4 | 723049 | bulk | Stroma_remaining | bulk | TMA1_Selection21 | TMA1 | Selection 21 | NaN | yes | GD_TMA1 | ... | 1E | S00102 | MMRd | negative | positive | HLA defect | mutation (exon 2) | B2M defect | TMA1_Selection21 | Stroma |
5 rows × 22 columns
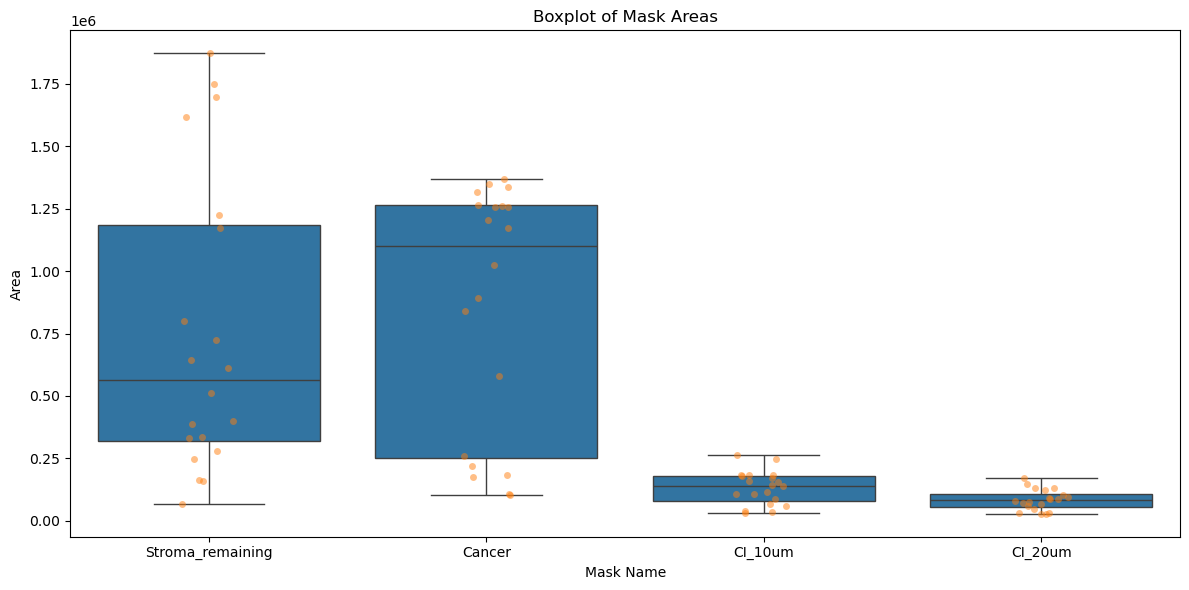
Areas of the mask
plt.figure(figsize=(12, 6))
sns.boxplot(x='mask_name', y='area', data=adata.obs, showfliers=False)
# Overlay the points
sns.stripplot(x='mask_name', y='area', data=adata.obs, alpha=0.5)
plt.title('Boxplot of Mask Areas')
plt.xlabel('Mask Name')
plt.ylabel('Area')
plt.tight_layout()
plt.show()
Normalization
We will normalize the values, as it was single cell data
# Normalize the counts in the 'X' matrix
adata.X = np.nan_to_num(adata.X / adata.obs['area'].values[:, None])*100
# sc.pp.scale(adata)
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.tl.pca(adata)
pct = adata.uns['pca']['variance_ratio'] / sum(adata.uns['pca']['variance_ratio']) * 100
cumu = np.cumsum(pct)
# Point 1.
co1 = list(np.where(np.logical_and(cumu > 90, pct < 5))[0])[0]
# Point 2.
x = list(np.where(pct[0:len(pct)-1] - pct[1:len(pct)] > 0.1)[0])
x.sort(reverse=True)
co2 = x[0]+1
# Elbow:
elbow = min(co1, co2) + 1 # Without the +1, it gives the index and not the PC number
# (indices in python start with 0, unlike R that start with 1)
print('Elbow at: ', elbow)
sc.pl.pca_variance_ratio(adata, n_pcs=50, log=True)
sc.pp.neighbors(adata, n_neighbors=20) #, n_pcs=elbow)
sc.tl.umap(adata)
sc.pl.umap(
adata,
color= ["mask_name", "selection"],
size=15,
)
Elbow at: 15
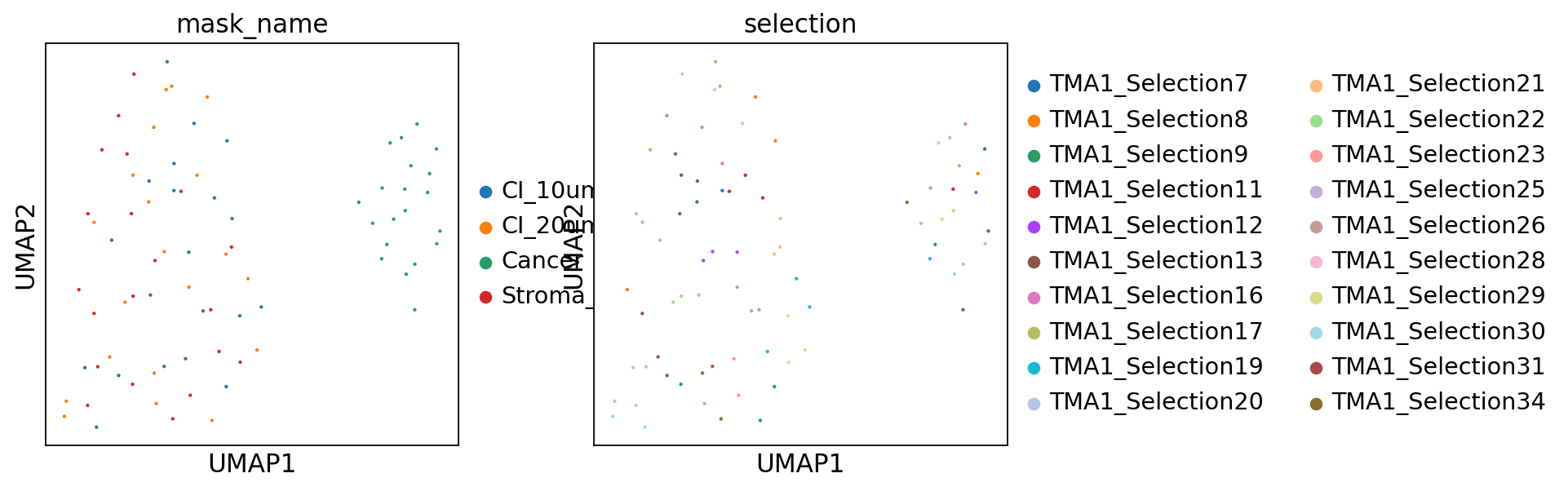
Cancer masks are clearly distinct from Stroma masks, which it is to be expected. From the UMAP there is no clear separation between Stroma and interface masks.
Differential gene expression
Stroma vs Interface vs Cancer
Stroma vs Interface
Interface vs cancer
# function for rank genes and derive dfs
def rank_subgroup_results_dotplot(groupcl, column_name = 'Final group', save_fig=None):
# groupcl.uns['log1p']['base'] = None
sc.tl.rank_genes_groups(groupcl,column_name, method='wilcoxon', pts=True)
sc.tl.filter_rank_genes_groups(groupcl, groupby=column_name, min_fold_change=1)
# Extract the results
result = groupcl.uns['rank_genes_groups']
groups = result['names'].dtype.names
# Create a DataFrame to store the results
result_df = pd.DataFrame()
for group in groups:
group_df = pd.DataFrame({
'gene': result['names'][group],
'logfoldchanges': result['logfoldchanges'][group],
'pvals': result['pvals'][group],
'pvals_adj': result['pvals_adj'][group],
'scores': result['scores'][group]
})
group_df['mask'] = group
result_df = pd.concat([result_df, group_df], ignore_index=True)
if save_fig:
sc.pl.rank_genes_groups_dotplot(groupcl, groupby=column_name, standard_scale="var", n_genes=15,save='dotplot.png')
else:
sc.pl.rank_genes_groups_dotplot(groupcl, groupby=column_name, standard_scale="var", n_genes=15)
return result_df
column_name = 'mask_name_combined'
groupcl = adata
groupcl.obs[column_name] = pd.Categorical(
groupcl.obs[column_name],
categories=['Stroma','Interface','Cancer',],
ordered=True
)
# groupcl.uns['log1p']['base'] = None
sc.tl.rank_genes_groups(groupcl,column_name, method='wilcoxon', pts=True)
sc.tl.filter_rank_genes_groups(groupcl, groupby=column_name, min_fold_change=1)
# Extract the results
result = groupcl.uns['rank_genes_groups']
groups = result['names'].dtype.names
# Create a DataFrame to store the results
results_df = pd.DataFrame()
for group in groups:
group_df = pd.DataFrame({
'gene': result['names'][group],
'logfoldchanges': result['logfoldchanges'][group],
'pvals': result['pvals'][group],
'pvals_adj': result['pvals_adj'][group],
'scores': result['scores'][group]
})
group_df['mask'] = group
results_df = pd.concat([results_df, group_df], ignore_index=True)
sc.set_figure_params(scanpy=True, fontsize=14)
dotplot = sc.pl.rank_genes_groups_dotplot(
groupcl,
groupby=column_name,
standard_scale="var",
n_genes=15, #15
dendrogram=True,
return_fig=True,
# swap_axes=True,
)
dotplot.savefig('figures/dotplot_highres.png', dpi=500, bbox_inches='tight')
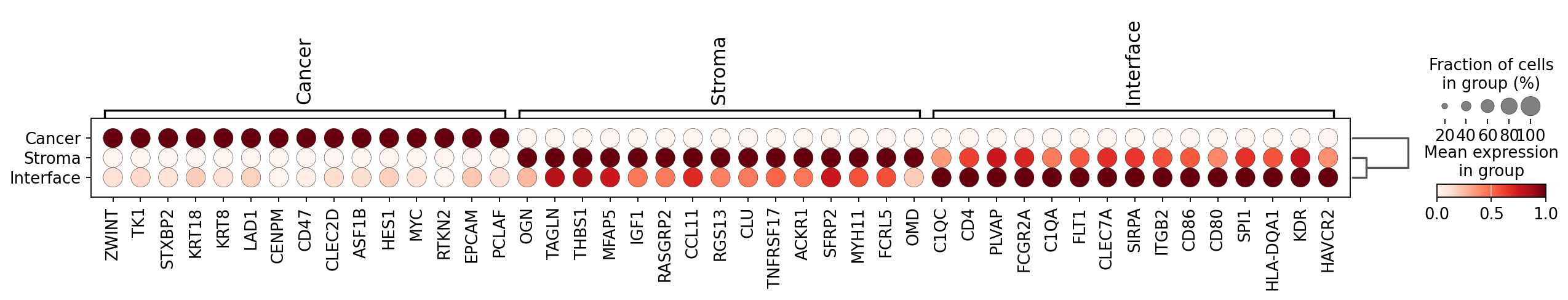
results_df
| gene | logfoldchanges | pvals | pvals_adj | scores | mask | |
|---|---|---|---|---|---|---|
| 0 | OGN | 1.377607 | 1.707981e-05 | 9.676217e-04 | 4.300000 | Stroma |
| 1 | TAGLN | 1.534681 | 5.244734e-05 | 1.798195e-03 | 4.044445 | Stroma |
| 2 | THBS1 | 1.016705 | 1.383436e-04 | 3.320246e-03 | 3.811111 | Stroma |
| 3 | MFAP5 | 1.286742 | 2.797832e-04 | 5.371837e-03 | 3.633333 | Stroma |
| 4 | IGF1 | 1.382461 | 2.797832e-04 | 5.371837e-03 | 3.633333 | Stroma |
| ... | ... | ... | ... | ... | ... | ... |
| 1435 | COL4A2 | -2.389624 | 2.616785e-11 | 5.410896e-10 | -6.666667 | Cancer |
| 1436 | LUM | -2.758327 | 2.616785e-11 | 5.410896e-10 | -6.666667 | Cancer |
| 1437 | TAGLN | -2.997534 | 2.616785e-11 | 5.410896e-10 | -6.666667 | Cancer |
| 1438 | VIM | -2.002866 | 2.616785e-11 | 5.410896e-10 | -6.666667 | Cancer |
| 1439 | LGALS1 | -2.143261 | 2.616785e-11 | 5.410896e-10 | -6.666667 | Cancer |
1440 rows × 6 columns
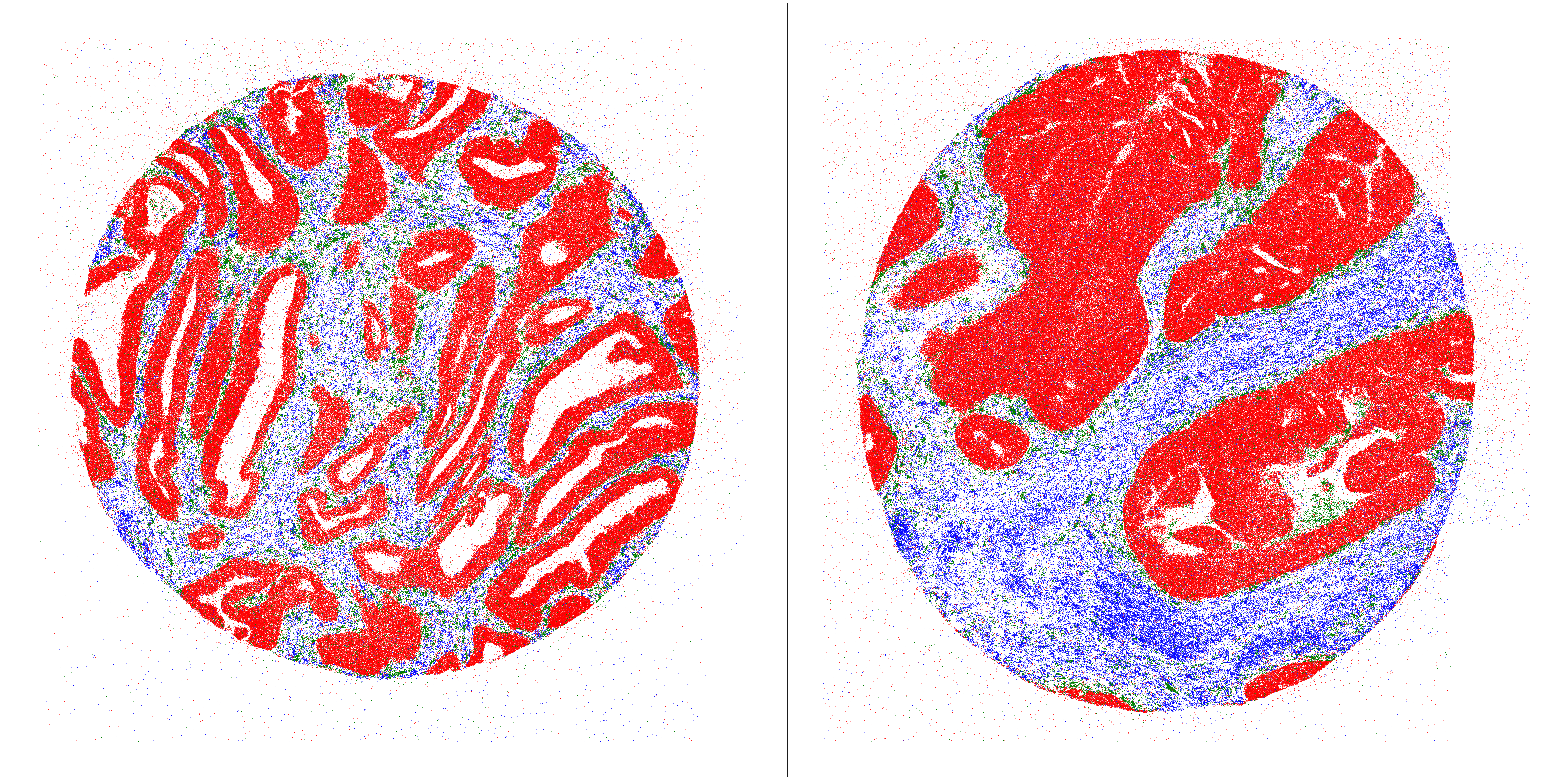
Plot genes back to 2 core samples
s_genes = results_df[results_df['mask']=='Stroma'].sort_values(by='scores', ascending=False)['gene'][:10]
t_genes = results_df[results_df['mask']=='Cancer'].sort_values(by='scores', ascending=False)['gene'][:10]
tb_genes = results_df[results_df['mask']=='Interface'].sort_values(by='scores', ascending=False)['gene'][:10]
fig1 = '../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection30_filtered.csv'
fig2 = '../../xenium_data/HLA/GD_TMA1_S3/fov_filtered/TMA1_Selection13_filtered.csv'
df1 = pd.read_csv(fig1)
df1 = df1[['x_location', 'y_location', 'feature_name']]
df1 = df1.rename(columns={'feature_name': 'target'})
df1['X'] = df1['x_location'] - min(df1['x_location'])
df1['Y'] = df1['y_location'] - min(df1['y_location'])
height1 = int(max(df1['X'])) + 1
width1 = int(max(df1['Y'])) + 1
df2 = pd.read_csv(fig2)
df2 = df2[['x_location', 'y_location', 'feature_name']]
df2 = df2.rename(columns={'feature_name': 'target'})
df2['X'] = df2['x_location'] - min(df2['x_location'])
df2['Y'] = df2['y_location'] - min(df2['y_location'])
height2 = int(max(df2['X'])) + 1
width2 = int(max(df2['Y'])) + 1
def plot_data(ax, df, s_genes, t_genes, tb_genes):
s_df = df[df['target'].isin(s_genes)]
ax.scatter(s_df['X'], s_df['Y'],
c='blue',
s=1, # Default size for blue
alpha=0.8, # Default alpha for blue
label='Stroma' # Add label for legend
)
t_df = df[df['target'].isin(t_genes)]
ax.scatter(t_df['X'], t_df['Y'],
c='red',
s=1, # Default size for blue
alpha=0.8,
label='Cancer' # Add label for legend
)
ci_df = df[df['target'].isin(tb_genes)]
ax.scatter(ci_df['X'], ci_df['Y'],
c='green',
s=1,
alpha=1.0,
label='Cancer Interface' # Add label for legend
)
ax.grid(False)
# Remove x and y tick labels and marks
ax.set_xticks([])
ax.set_yticks([])
ax.tick_params(left=False, bottom=False)
# Set up the figure and axes
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(50, 25))
# Plot data for both dataframes
plot_data(ax2, df1, list(s_genes), list(t_genes), list(tb_genes))
plot_data(ax1, df2,list(s_genes), list(t_genes), list(tb_genes))
# Adjust layout and display the plot
plt.tight_layout()
plt.show()
extent1 = ax1.get_window_extent().transformed(fig.dpi_scale_trans.inverted())
fig.savefig('figures/plot1.png', bbox_inches=extent1, dpi=300)
# Save the second subplot
extent2 = ax2.get_window_extent().transformed(fig.dpi_scale_trans.inverted())
fig.savefig('figures/plot2.png', bbox_inches=extent2, dpi=300)
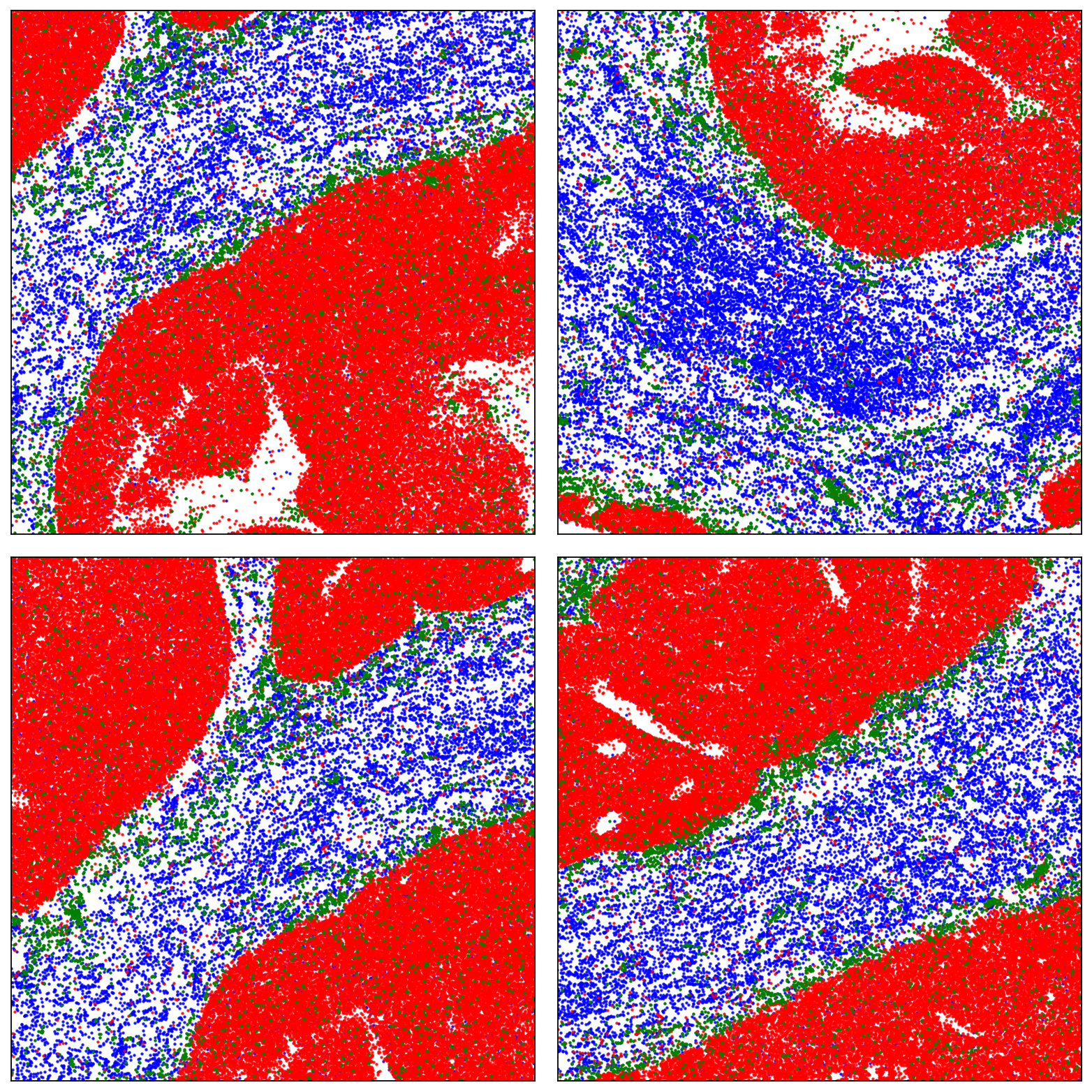
import math
zoom_centers = [
(1000, 800),
(900,350),
(900,900),
(1350,1150)
]
half_width = 250
half_height = 250
n_zooms = len(zoom_centers)
n_cols = 2
n_rows = math.ceil(n_zooms / n_cols)
fig, axes = plt.subplots(n_rows, n_cols, figsize=(5 * n_cols, 5 * n_rows))
axes = axes.flatten()
for i, (center_x, center_y) in enumerate(zoom_centers):
ax = axes[i]
plot_data(ax, df1, list(s_genes), list(t_genes), list(tb_genes))
ax.set_xlim(center_x - half_width, center_x + half_width)
ax.set_ylim(center_y - half_height, center_y + half_height)
# ax.set_title(f'Zoom {i+1}: Center ({center_x}, {center_y})')
# Save each individual axis to its own figure
single_fig = plt.figure()
single_ax = single_fig.add_subplot(111)
plot_data(single_ax, df1, list(s_genes), list(t_genes), list(tb_genes))
single_ax.set_xlim(center_x - half_width, center_x + half_width)
single_ax.set_ylim(center_y - half_height, center_y + half_height)
single_ax.axis('off')
single_fig.savefig(f'figures/zoom_section_{i+1}.png', dpi=300, bbox_inches='tight', pad_inches=0)
plt.close(single_fig) # Close to avoid clutter
# Hide extra unused axes
for j in range(i + 1, len(axes)):
axes[j].axis('off')
plt.tight_layout()
plt.show()
zoom_centers = [
(700, 500), # center 1
(1000, 800), # center 2
(1200, 1200), # center 3
(1300,650),
(1300,850),
(1000,750)
]
half_width = 250
half_height = 250
n_zooms = len(zoom_centers)
n_cols = 2
n_rows = math.ceil(n_zooms / n_cols)
fig, axes = plt.subplots(n_rows, n_cols, figsize=(5 * n_cols, 5 * n_rows))
axes = axes.flatten()
for i, (center_x, center_y) in enumerate(zoom_centers):
ax = axes[i]
plot_data(ax, df2, list(s_genes), list(t_genes), list(tb_genes))
ax.set_xlim(center_x - half_width, center_x + half_width)
ax.set_ylim(center_y - half_height, center_y + half_height)
# ax.set_title(f'Zoom {i+1}: Center ({center_x}, {center_y})')
# Save each individual axis to its own figure
single_fig = plt.figure()
single_ax = single_fig.add_subplot(111)
plot_data(single_ax, df2, list(s_genes), list(t_genes), list(tb_genes))
single_ax.set_xlim(center_x - half_width, center_x + half_width)
single_ax.set_ylim(center_y - half_height, center_y + half_height)
single_ax.axis('off')
single_fig.savefig(f'figures/2_zoom_section_{i+1}.png', dpi=300, bbox_inches='tight', pad_inches=0)
plt.close(single_fig) # Close to avoid clutter
# Hide extra unused axes
for j in range(i + 1, len(axes)):
axes[j].axis('off')
plt.tight_layout()
plt.show()
